 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA new correlation clustering method for cancer mutation analysis
Paper and Code
Jan 25, 2016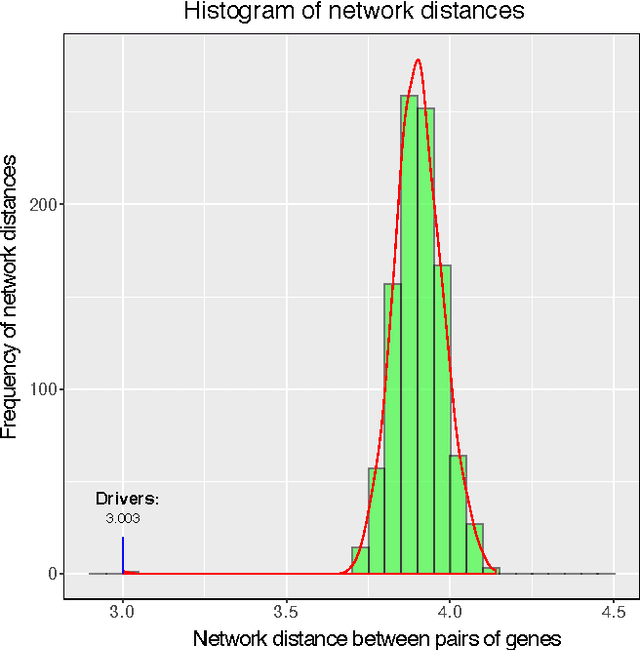
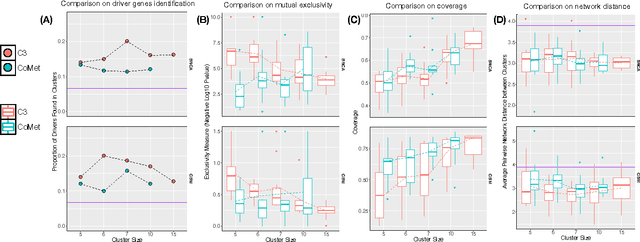
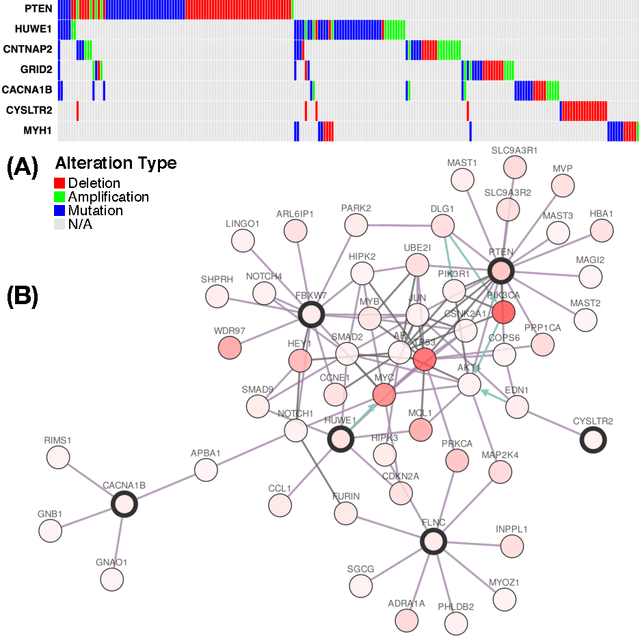
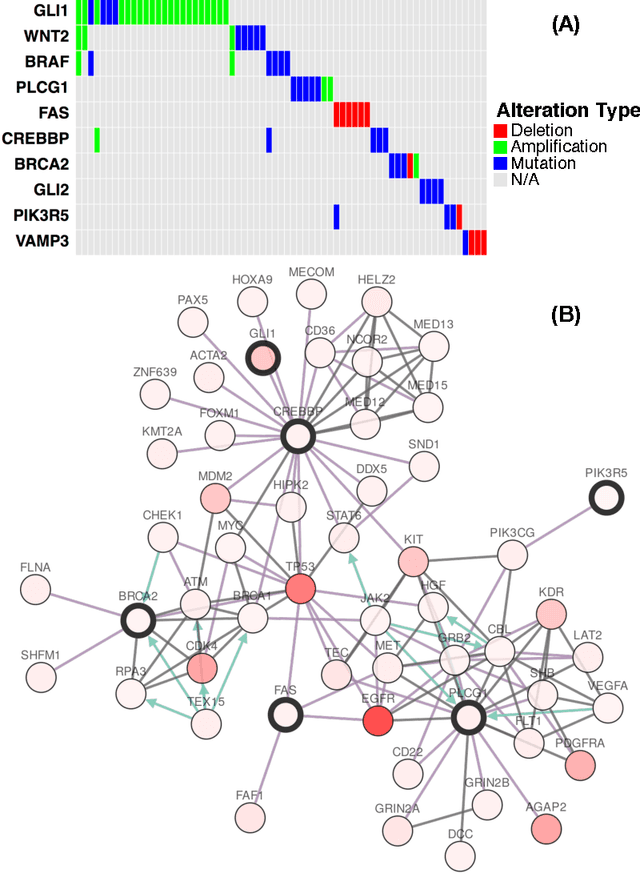
Cancer genomes exhibit a large number of different alterations that affect many genes in a diverse manner. It is widely believed that these alterations follow combinatorial patterns that have a strong connection with the underlying molecular interaction networks and functional pathways. A better understanding of the generative mechanisms behind the mutation rules and their influence on gene communities is of great importance for the process of driver mutations discovery and for identification of network modules related to cancer development and progression. We developed a new method for cancer mutation pattern analysis based on a constrained form of correlation clustering. Correlation clustering is an agnostic learning method that can be used for general community detection problems in which the number of communities or their structure is not known beforehand. The resulting algorithm, named $C^3$, leverages mutual exclusivity of mutations, patient coverage, and driver network concentration principles; it accepts as its input a user determined combination of heterogeneous patient data, such as that available from TCGA (including mutation, copy number, and gene expression information), and creates a large number of clusters containing mutually exclusive mutated genes in a particular type of cancer. The cluster sizes may be required to obey some useful soft size constraints, without impacting the computational complexity of the algorithm. To test $C^3$, we performed a detailed analysis on TCGA breast cancer and glioblastoma data and showed that our algorithm outperforms the state-of-the-art CoMEt method in terms of discovering mutually exclusive gene modules and identifying driver genes. Our $C^3$ method represents a unique tool for efficient and reliable identification of mutation patterns and driver pathways in large-scale cancer genomics studies.