 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAt FullTilt: Real-Time Open-Set 3D Macromolecule Detection Directly from Tilted 2D Projections
Apr 12, 2026Open-set 3D macromolecule detection in cryogenic electron tomography eliminates the need for target-specific model retraining. However, strict VRAM constraints prohibit processing an entire 3D tomogram, forcing current methods to rely on slow sliding-window inference over extracted subvolumes. To overcome this, we propose FullTilt, an end-to-end framework that redefines 3D detection by operating directly on aligned 2D tilt-series. Because a tilt-series contains significantly fewer images than slices in a reconstructed tomogram, FullTilt eliminates redundant volumetric computation, accelerating inference by orders of magnitude. To process the entire tilt-series simultaneously, we introduce a tilt-series encoder to efficiently fuse cross-view information. We further propose a multiclass visual prompt encoder for flexible prompting, a tilt-aware query initializer to effectively anchor 3D queries, and an auxiliary geometric primitives module to enhance the model's understanding of multi-view geometry while improving robustness to adverse imaging artifacts. Extensive evaluations on three real-world datasets demonstrate that FullTilt achieves state-of-the-art zero-shot performance while drastically reducing runtime and VRAM requirements, paving the way for rapid, large-scale visual proteomics analysis. All code and data will be publicly available upon publication.
Cryo-ZSSR: multiple-image super-resolution based on deep internal learning
Nov 22, 2020
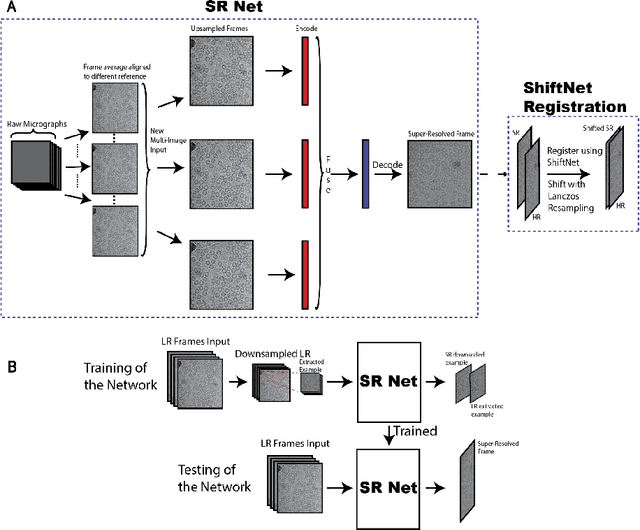
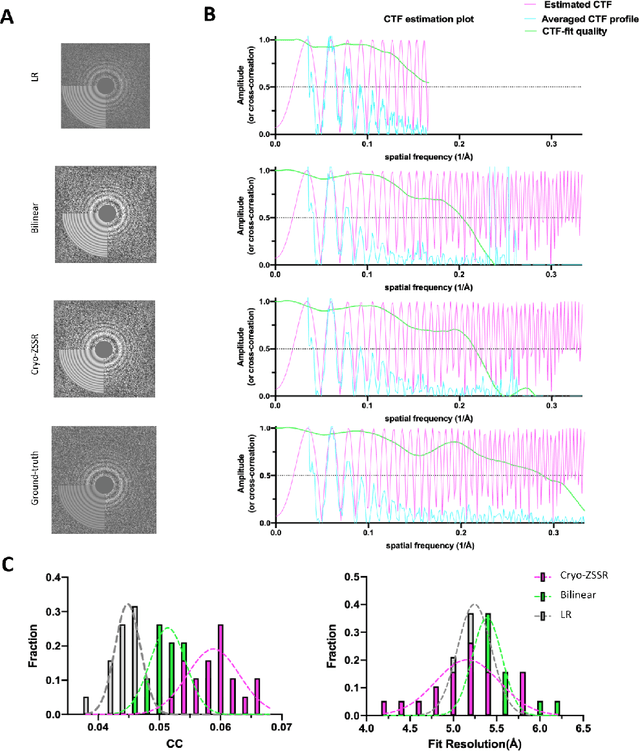
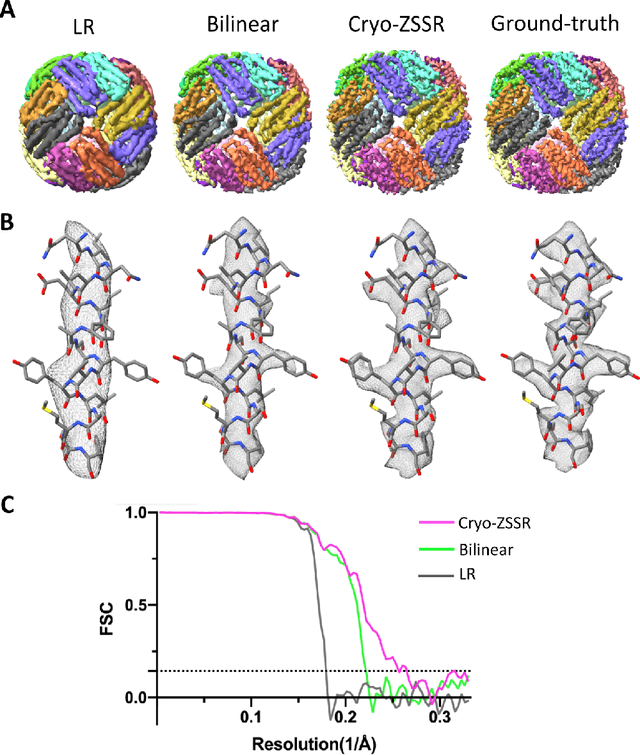
Single-particle cryo-electron microscopy (cryo-EM) is an emerging imaging modality capable of visualizing proteins and macro-molecular complexes at near-atomic resolution. The low electron-doses used to prevent sample radiation damage, result in images where the power of the noise is 100 times greater than the power of the signal. To overcome the low-SNRs, hundreds of thousands of particle projections acquired over several days of data collection are averaged in 3D to determine the structure of interest. Meanwhile, recent image super-resolution (SR) techniques based on neural networks have shown state of the art performance on natural images. Building on these advances, we present a multiple-image SR algorithm based on deep internal learning designed specifically to work under low-SNR conditions. Our approach leverages the internal image statistics of cryo-EM movies and does not require training on ground-truth data. When applied to a single-particle dataset of apoferritin, we show that the resolution of 3D structures obtained from SR micrographs can surpass the limits imposed by the imaging system. Our results indicate that the combination of low magnification imaging with image SR has the potential to accelerate cryo-EM data collection without sacrificing resolution.
Unsupervised particle sorting for high-resolution single-particle cryo-EM
Oct 22, 2019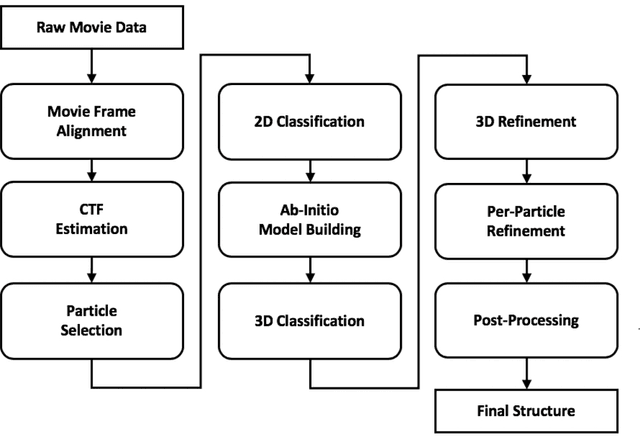
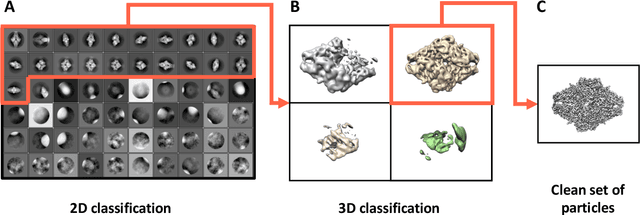
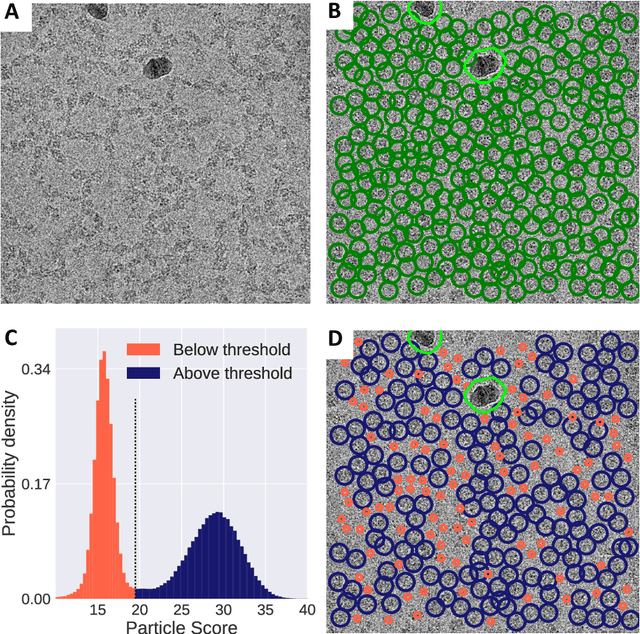
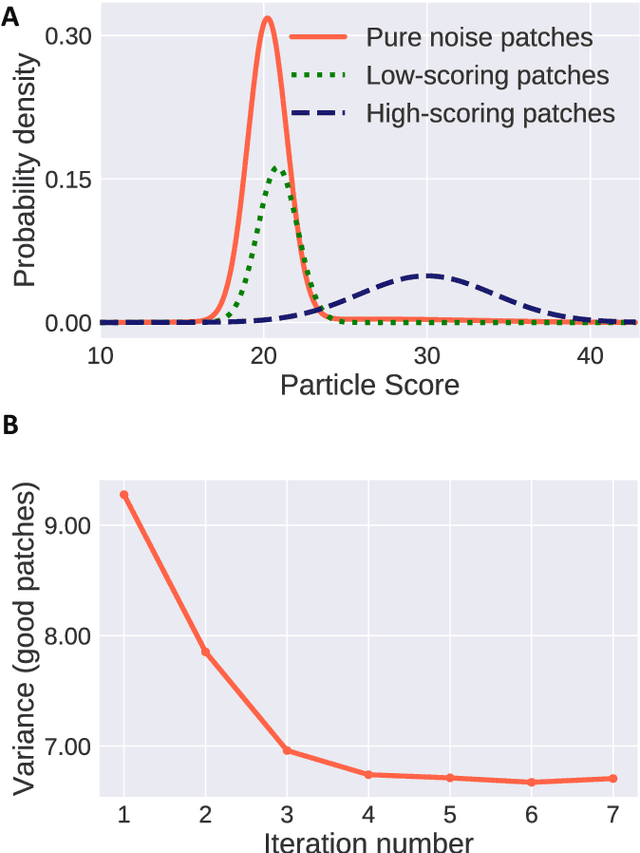
Single-particle cryo-Electron Microscopy (EM) has become a popular technique for determining the structure of challenging biomolecules that are inaccessible to other technologies. Recent advances in automation, both in data collection and data processing, have significantly lowered the barrier for non-expert users to successfully execute the structure determination workflow. Many critical data processing steps, however, still require expert user intervention in order to converge to the correct high-resolution structure. In particular, strategies to identify homogeneous populations of particles rely heavily on subjective criteria that are not always consistent or reproducible among different users. Here, we explore the use of unsupervised strategies for particle sorting that are compatible with the autonomous operation of the image processing pipeline. More specifically, we show that particles can be successfully sorted based on a simple statistical model for the distribution of scores assigned during refinement. This represents an important step towards the development of automated workflows for protein structure determination using single-particle cryo-EM.
Fundamental Limits in Multi-image Alignment
Feb 04, 2016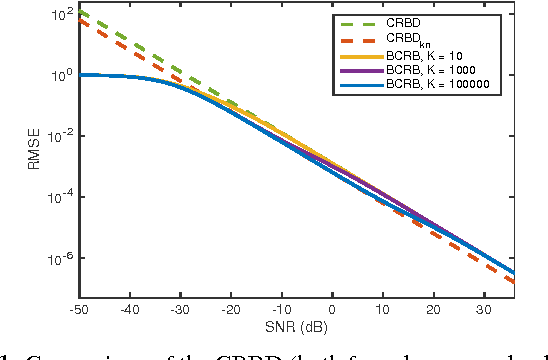
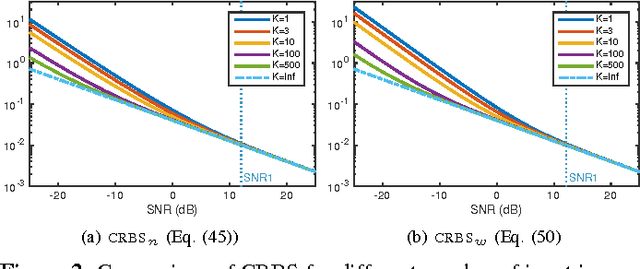

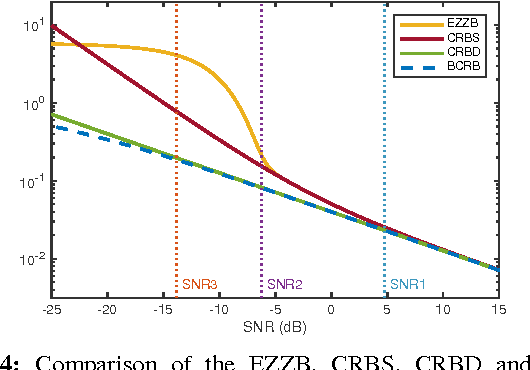
The performance of multi-image alignment, bringing different images into one coordinate system, is critical in many applications with varied signal-to-noise ratio (SNR) conditions. A great amount of effort is being invested into developing methods to solve this problem. Several important questions thus arise, including: Which are the fundamental limits in multi-image alignment performance? Does having access to more images improve the alignment? Theoretical bounds provide a fundamental benchmark to compare methods and can help establish whether improvements can be made. In this work, we tackle the problem of finding the performance limits in image registration when multiple shifted and noisy observations are available. We derive and analyze the Cram\'er-Rao and Ziv-Zakai lower bounds under different statistical models for the underlying image. The accuracy of the derived bounds is experimentally assessed through a comparison to the maximum likelihood estimator. We show the existence of different behavior zones depending on the difficulty level of the problem, given by the SNR conditions of the input images. We find that increasing the number of images is only useful below a certain SNR threshold, above which the pairwise MLE estimation proves to be optimal. The analysis we present here brings further insight into the fundamental limitations of the multi-image alignment problem.