 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConformalHDC: Uncertainty-Aware Hyperdimensional Computing with Application to Neural Decoding
Feb 24, 2026Hyperdimensional Computing (HDC) offers a computationally efficient paradigm for neuromorphic learning. Yet, it lacks rigorous uncertainty quantification, leading to open decision boundaries and, consequently, vulnerability to outliers, adversarial perturbations, and out-of-distribution inputs. To address these limitations, we introduce ConformalHDC, a unified framework that combines the statistical guarantees of conformal prediction with the computational efficiency of HDC. For this framework, we propose two complementary variations. First, the set-valued formulation provides finite-sample, distribution-free coverage guarantees. Using carefully designed conformity scores, it forms enclosed decision boundaries that improve robustness to non-conforming inputs. Second, the point-valued formulation leverages the same conformity scores to produce a single prediction when desired, potentially improving accuracy over traditional HDC by accounting for class interactions. We demonstrate the broad applicability of the proposed framework through evaluations on multiple real-world datasets. In particular, we apply our method to the challenging problem of decoding non-spatial stimulus information from the spiking activity of hippocampal neurons recorded as subjects performed a sequence memory task. Our results show that ConformalHDC not only accurately decodes the stimulus information represented in the neural activity data, but also provides rigorous uncertainty estimates and correctly abstains when presented with data from other behavioral states. Overall, these capabilities position the framework as a reliable, uncertainty-aware foundation for neuromorphic computing.
ERNIE 5.0 Technical Report
Feb 04, 2026In this report, we introduce ERNIE 5.0, a natively autoregressive foundation model desinged for unified multimodal understanding and generation across text, image, video, and audio. All modalities are trained from scratch under a unified next-group-of-tokens prediction objective, based on an ultra-sparse mixture-of-experts (MoE) architecture with modality-agnostic expert routing. To address practical challenges in large-scale deployment under diverse resource constraints, ERNIE 5.0 adopts a novel elastic training paradigm. Within a single pre-training run, the model learns a family of sub-models with varying depths, expert capacities, and routing sparsity, enabling flexible trade-offs among performance, model size, and inference latency in memory- or time-constrained scenarios. Moreover, we systematically address the challenges of scaling reinforcement learning to unified foundation models, thereby guaranteeing efficient and stable post-training under ultra-sparse MoE architectures and diverse multimodal settings. Extensive experiments demonstrate that ERNIE 5.0 achieves strong and balanced performance across multiple modalities. To the best of our knowledge, among publicly disclosed models, ERNIE 5.0 represents the first production-scale realization of a trillion-parameter unified autoregressive model that supports both multimodal understanding and generation. To facilitate further research, we present detailed visualizations of modality-agnostic expert routing in the unified model, alongside comprehensive empirical analysis of elastic training, aiming to offer profound insights to the community.
Heterogeneous Graph Alignment for Joint Reasoning and Interpretability
Jan 30, 2026Multi-graph learning is crucial for extracting meaningful signals from collections of heterogeneous graphs. However, effectively integrating information across graphs with differing topologies, scales, and semantics, often in the absence of shared node identities, remains a significant challenge. We present the Multi-Graph Meta-Transformer (MGMT), a unified, scalable, and interpretable framework for cross-graph learning. MGMT first applies Graph Transformer encoders to each graph, mapping structure and attributes into a shared latent space. It then selects task-relevant supernodes via attention and builds a meta-graph that connects functionally aligned supernodes across graphs using similarity in the latent space. Additional Graph Transformer layers on this meta-graph enable joint reasoning over intra- and inter-graph structure. The meta-graph provides built-in interpretability: supernodes and superedges highlight influential substructures and cross-graph alignments. Evaluating MGMT on both synthetic datasets and real-world neuroscience applications, we show that MGMT consistently outperforms existing state-of-the-art models in graph-level prediction tasks while offering interpretable representations that facilitate scientific discoveries. Our work establishes MGMT as a unified framework for structured multi-graph learning, advancing representation techniques in domains where graph-based data plays a central role.
Meta Fusion: A Unified Framework For Multimodality Fusion with Mutual Learning
Jul 27, 2025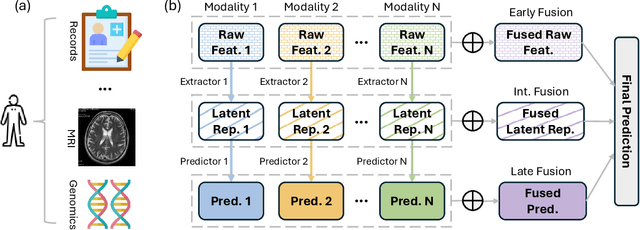

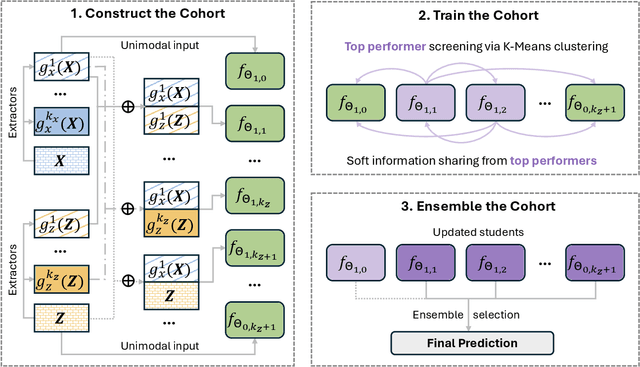

Developing effective multimodal data fusion strategies has become increasingly essential for improving the predictive power of statistical machine learning methods across a wide range of applications, from autonomous driving to medical diagnosis. Traditional fusion methods, including early, intermediate, and late fusion, integrate data at different stages, each offering distinct advantages and limitations. In this paper, we introduce Meta Fusion, a flexible and principled framework that unifies these existing strategies as special cases. Motivated by deep mutual learning and ensemble learning, Meta Fusion constructs a cohort of models based on various combinations of latent representations across modalities, and further boosts predictive performance through soft information sharing within the cohort. Our approach is model-agnostic in learning the latent representations, allowing it to flexibly adapt to the unique characteristics of each modality. Theoretically, our soft information sharing mechanism reduces the generalization error. Empirically, Meta Fusion consistently outperforms conventional fusion strategies in extensive simulation studies. We further validate our approach on real-world applications, including Alzheimer's disease detection and neural decoding.
Structured Conformal Inference for Matrix Completion with Applications to Group Recommender Systems
Apr 26, 2024



We develop a conformal inference method to construct joint confidence regions for structured groups of missing entries within a sparsely observed matrix. This method is useful to provide reliable uncertainty estimation for group-level collaborative filtering; for example, it can be applied to help suggest a movie for a group of friends to watch together. Unlike standard conformal techniques, which make inferences for one individual at a time, our method achieves stronger group-level guarantees by carefully assembling a structured calibration data set mimicking the patterns expected among the test group of interest. We propose a generalized weighted conformalization framework to deal with the lack of exchangeability arising from such structured calibration, and in this process we introduce several innovations to overcome computational challenges. The practicality and effectiveness of our method are demonstrated through extensive numerical experiments and an analysis of the MovieLens 100K data set.
Conformal inference is (almost) free for neural networks trained with early stopping
Jan 27, 2023



Early stopping based on hold-out data is a popular regularization technique designed to mitigate overfitting and increase the predictive accuracy of neural networks. Models trained with early stopping often provide relatively accurate predictions, but they generally still lack precise statistical guarantees unless they are further calibrated using independent hold-out data. This paper addresses the above limitation with conformalized early stopping: a novel method that combines early stopping with conformal calibration while efficiently recycling the same hold-out data. This leads to models that are both accurate and able to provide exact predictive inferences without multiple data splits nor overly conservative adjustments. Practical implementations are developed for different learning tasks -- outlier detection, multi-class classification, regression -- and their competitive performance is demonstrated on real data.
Integrative conformal p-values for powerful out-of-distribution testing with labeled outliers
Aug 23, 2022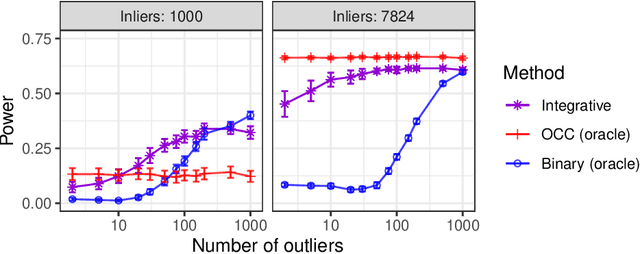
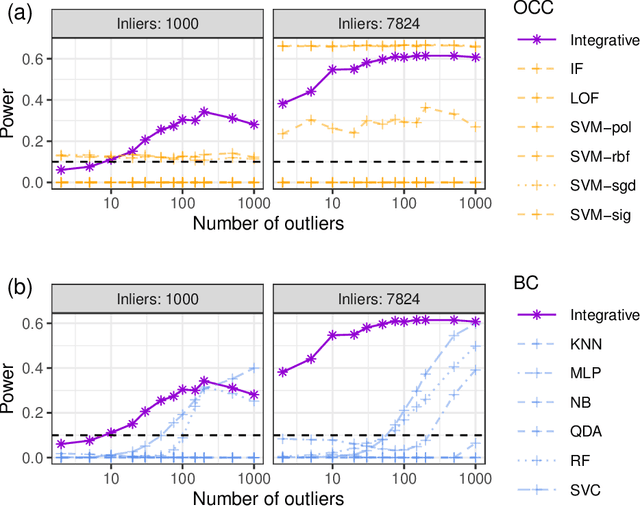
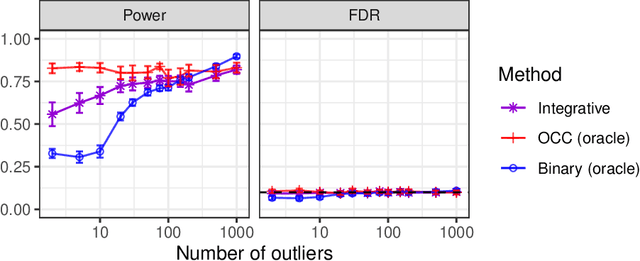
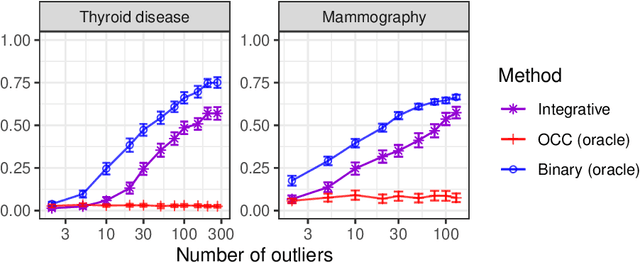
This paper develops novel conformal methods to test whether a new observation was sampled from the same distribution as a reference set. Blending inductive and transductive conformal inference in an innovative way, the described methods can re-weight standard conformal p-values based on dependent side information from known out-of-distribution data in a principled way, and can automatically take advantage of the most powerful model from any collection of one-class and binary classifiers. The solution can be implemented either through sample splitting or via a novel transductive cross-validation+ scheme which may also be useful in other applications of conformal inference, due to tighter guarantees compared to existing cross-validation approaches. After studying false discovery rate control and power within a multiple testing framework with several possible outliers, the proposed solution is shown to outperform standard conformal p-values through simulations as well as applications to image recognition and tabular data.
Locally Adaptive Transfer Learning Algorithms for Large-Scale Multiple Testing
Mar 25, 2022


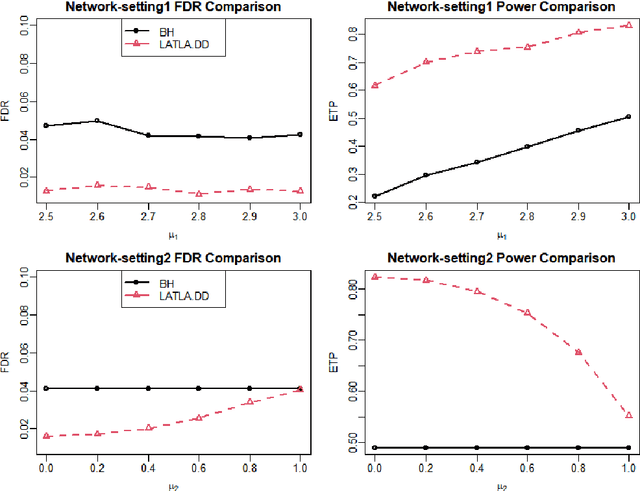
Transfer learning has enjoyed increasing popularity in a range of big data applications. In the context of large-scale multiple testing, the goal is to extract and transfer knowledge learned from related source domains to improve the accuracy of simultaneously testing of a large number of hypotheses in the target domain. This article develops a locally adaptive transfer learning algorithm (LATLA) for transfer learning for multiple testing. In contrast with existing covariate-assisted multiple testing methods that require the auxiliary covariates to be collected alongside the primary data on the same testing units, LATLA provides a principled and generic transfer learning framework that is capable of incorporating multiple samples of auxiliary data from related source domains, possibly in different dimensions/structures and from diverse populations. Both the theoretical and numerical results show that LATLA controls the false discovery rate and outperforms existing methods in power. LATLA is illustrated through an application to genome-wide association studies for the identification of disease-associated SNPs by cross-utilizing the auxiliary data from a related linkage analysis.