 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeZeoformer: Coarse-Grained Periodic Graph Transformer for OSDA-Zeolite Affinity Prediction
Aug 26, 2024To date, the International Zeolite Association Structure Commission (IZA-SC) has cataloged merely 255 distinct zeolite structures, with millions of theoretically possible structures yet to be discovered. The synthesis of a specific zeolite typically necessitates the use of an organic structure-directing agent (OSDA), since the selectivity for a particular zeolite is largely determined by the affinity between the OSDA and the zeolite. Therefore, finding the best affinity OSDA-zeolite pair is the key to the synthesis of targeted zeolite. However, OSDA-zeolite pairs frequently exhibit complex geometric structures, i.e., a complex crystal structure formed by a large number of atoms. Although some existing machine learning methods can represent the periodicity of crystals, they cannot accurately represent crystal structures with local variability. To address this issue, we propose a novel approach called Zeoformer, which can effectively represent coarse-grained crystal periodicity and fine-grained local variability. Zeoformer reconstructs the unit cell centered around each atom and encodes the pairwise distances between this central atom and other atoms within the reconstructed unit cell. The introduction of pairwise distances within the reconstructed unit cell more effectively represents the overall structure of the unit cell and the differences between different unit cells, enabling the model to more accurately and efficiently predict the properties of OSDA-zeolite pairs and general crystal structures. Through comprehensive evaluation, our Zeoformer model demonstrates the best performance on OSDA-zeolite pair datasets and two types of crystal material datasets.
Curvature-based Transformer for Molecular Property Prediction
Jul 25, 2023
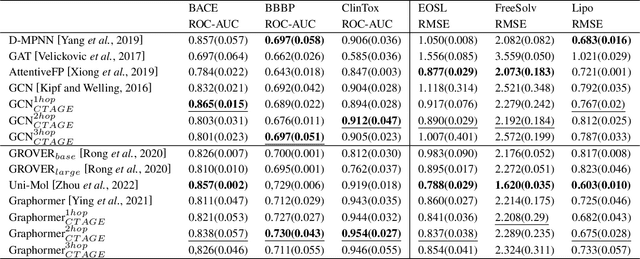


The prediction of molecular properties is one of the most important and challenging tasks in the field of artificial intelligence-based drug design. Among the current mainstream methods, the most commonly used feature representation for training DNN models is based on SMILES and molecular graphs, although these methods are concise and effective, they also limit the ability to capture spatial information. In this work, we propose Curvature-based Transformer to improve the ability of Graph Transformer neural network models to extract structural information on molecular graph data by introducing Discretization of Ricci Curvature. To embed the curvature in the model, we add the curvature information of the graph as positional Encoding to the node features during the attention-score calculation. This method can introduce curvature information from graph data without changing the original network architecture, and it has the potential to be extended to other models. We performed experiments on chemical molecular datasets including PCQM4M-LST, MoleculeNet and compared with models such as Uni-Mol, Graphormer, and the results show that this method can achieve the state-of-the-art results. It is proved that the discretized Ricci curvature also reflects the structural and functional relationship while describing the local geometry of the graph molecular data.