 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWaveFormer: Frequency-Time Decoupled Vision Modeling with Wave Equation
Jan 13, 2026Vision modeling has advanced rapidly with Transformers, whose attention mechanisms capture visual dependencies but lack a principled account of how semantic information propagates spatially. We revisit this problem from a wave-based perspective: feature maps are treated as spatial signals whose evolution over an internal propagation time (aligned with network depth) is governed by an underdamped wave equation. In this formulation, spatial frequency-from low-frequency global layout to high-frequency edges and textures-is modeled explicitly, and its interaction with propagation time is controlled rather than implicitly fixed. We derive a closed-form, frequency-time decoupled solution and implement it as the Wave Propagation Operator (WPO), a lightweight module that models global interactions in O(N log N) time-far lower than attention. Building on WPO, we propose a family of WaveFormer models as drop-in replacements for standard ViTs and CNNs, achieving competitive accuracy across image classification, object detection, and semantic segmentation, while delivering up to 1.6x higher throughput and 30% fewer FLOPs than attention-based alternatives. Furthermore, our results demonstrate that wave propagation introduces a complementary modeling bias to heat-based methods, effectively capturing both global coherence and high-frequency details essential for rich visual semantics. Codes are available at: https://github.com/ZishanShu/WaveFormer.
Deep manifold learning reveals hidden dynamics of proteasome autoregulation
Dec 23, 2020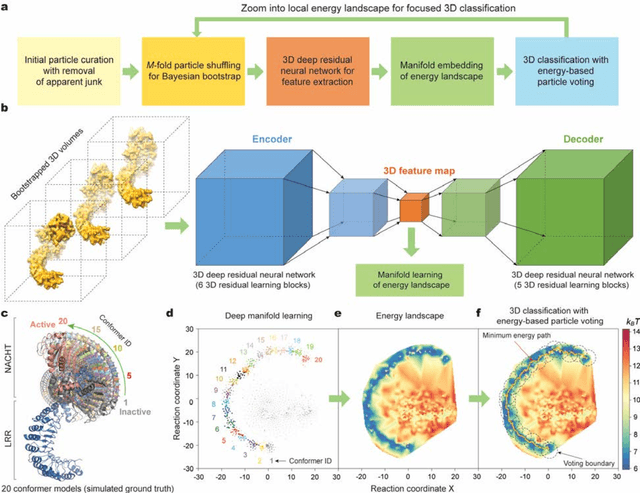
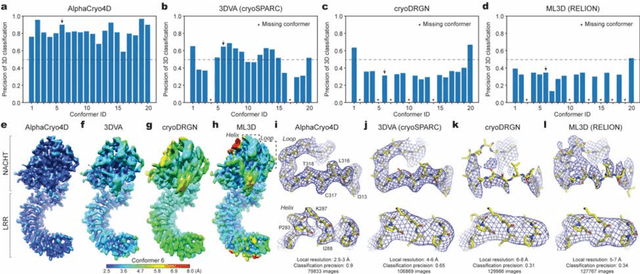
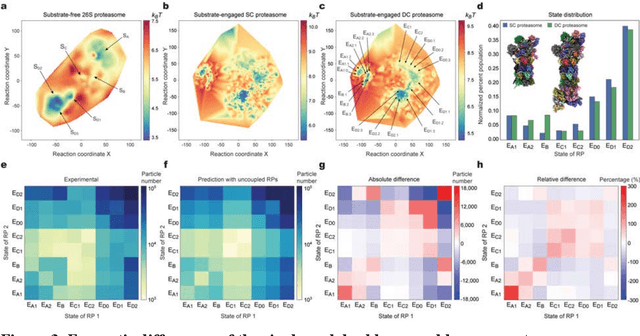
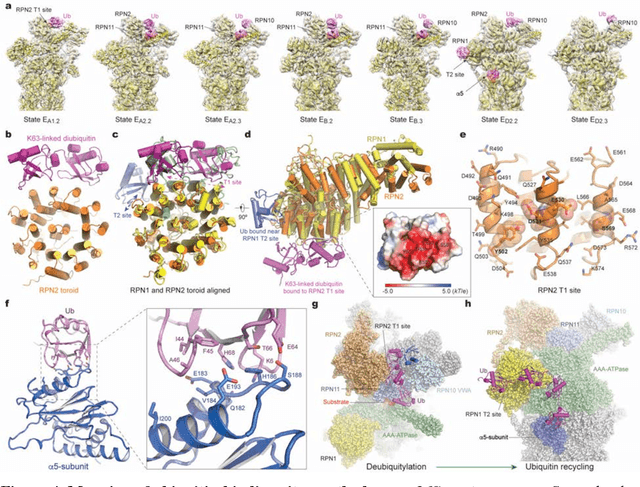
The 2.5-MDa 26S proteasome maintains proteostasis and regulates myriad cellular processes. How polyubiquitylated substrate interactions regulate proteasome activity is not understood. Here we introduce a deep manifold learning framework, named AlphaCryo4D, which enables atomic-level cryogenic electron microscopy (cryo-EM) reconstructions of nonequilibrium conformational continuum and reconstitutes hidden dynamics of proteasome autoregulation in the act of substrate degradation. AlphaCryo4D integrates 3D deep residual learning with manifold embedding of free-energy landscapes, which directs 3D clustering via an energy-based particle-voting algorithm. In blind assessments using simulated heterogeneous cryo-EM datasets, AlphaCryo4D achieved 3D classification accuracy three times that of conventional method and reconstructed continuous conformational changes of a 130-kDa protein at sub-3-angstrom resolution. By using AlphaCryo4D to analyze a single experimental cryo-EM dataset, we identified 64 conformers of the substrate-bound human 26S proteasome, revealing conformational entanglement of two regulatory particles in the doubly capped holoenzymes and their energetic differences with singly capped ones. Novel ubiquitin-binding sites are discovered on the RPN2, RPN10 and Alpha5 subunits to remodel polyubiquitin chains for deubiquitylation and recycle. Importantly, AlphaCryo4D choreographs single-nucleotide-exchange dynamics of proteasomal AAA-ATPase motor during translocation initiation, which upregulates proteolytic activity by allosterically promoting nucleophilic attack. Our systemic analysis illuminates a grand hierarchical allostery for proteasome autoregulation.
Unsupervised single-particle deep clustering via statistical manifold learning
Dec 31, 2016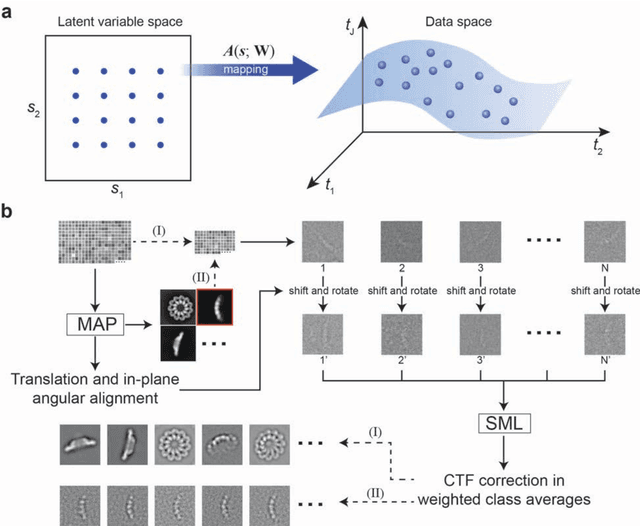
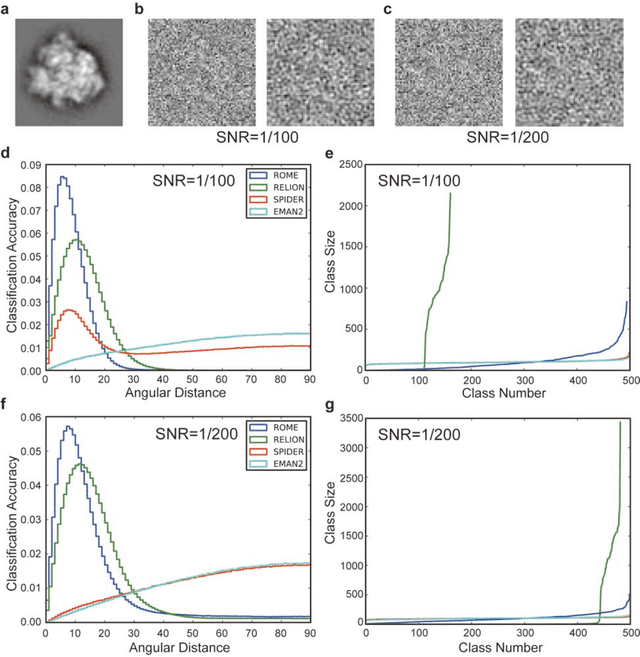
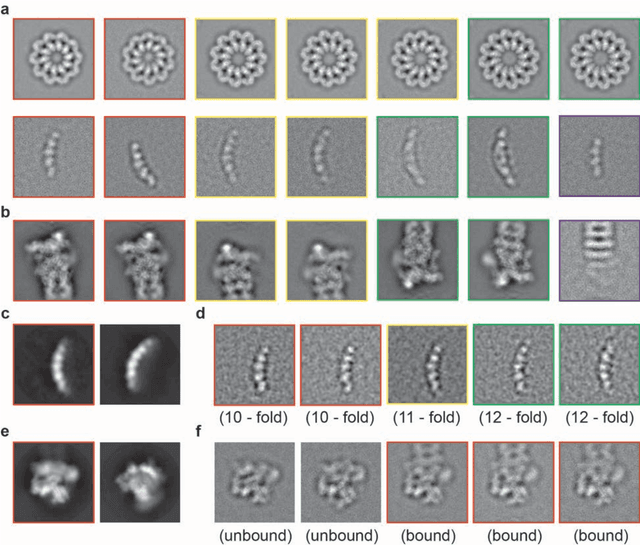
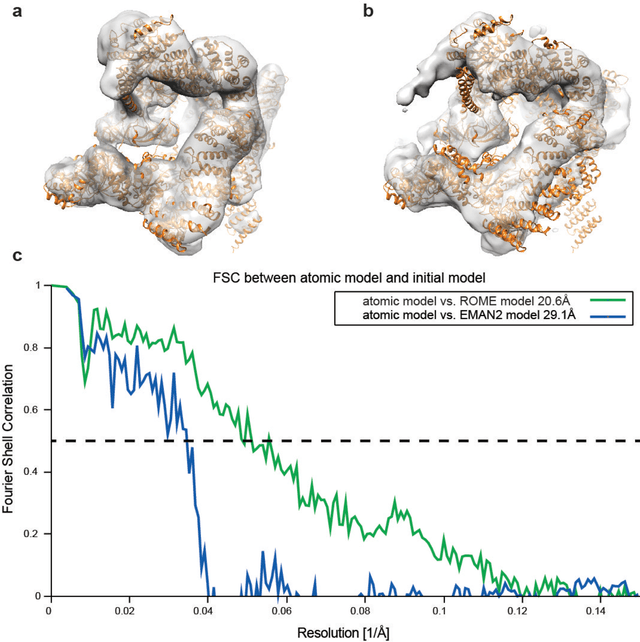
Motivation: Structural heterogeneity in single-particle cryo-electron microscopy (cryo-EM) data represents a major challenge for high-resolution structure determination. Unsupervised classification may serve as the first step in the assessment of structural heterogeneity. Traditional algorithms for unsupervised classification, such as K-means clustering and maximum likelihood optimization, may classify images into wrong classes with decreasing signal-to-noise-ratio (SNR) in the image data, yet demand increased cost in computation. Overcoming these limitations requires further development on clustering algorithms for high-performance cryo-EM data analysis. Results: Here we introduce a statistical manifold learning algorithm for unsupervised single-particle deep clustering. We show that statistical manifold learning improves classification accuracy by about 40% in the absence of input references for lower SNR data. Applications to several experimental datasets suggest that our deep clustering approach can detect subtle structural difference among classes. Through code optimization over the Intel high-performance computing (HPC) processors, our software implementation can generate thousands of reference-free class averages within several hours from hundreds of thousands of single-particle cryo-EM images, which allows significant improvement in ab initio 3D reconstruction resolution and quality. Our approach has been successfully applied in several structural determination projects. We expect that it provides a powerful computational tool in analyzing highly heterogeneous structural data and assisting in computational purification of single-particle datasets for high-resolution reconstruction.
* 29 pages, 5 figures
A deep convolutional neural network approach to single-particle recognition in cryo-electron microscopy
Dec 31, 2016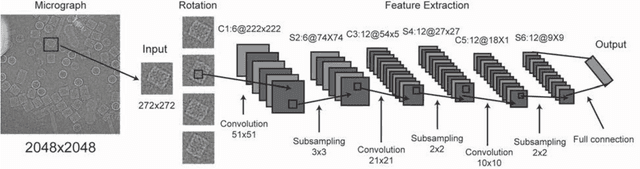
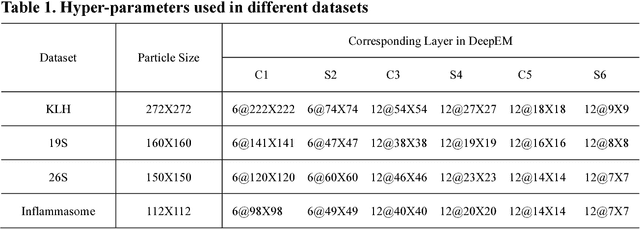
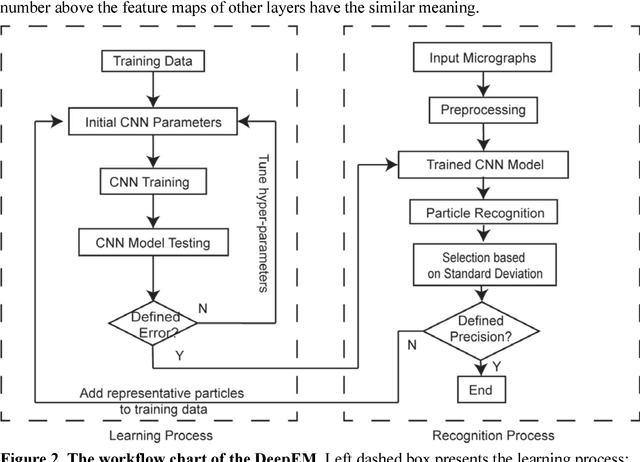
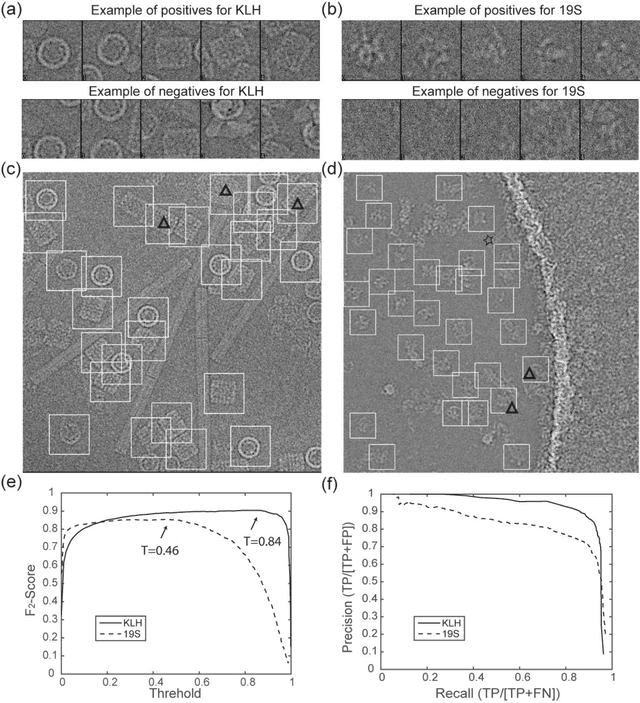
Background: Single-particle cryo-electron microscopy (cryo-EM) has become a popular tool for structural determination of biological macromolecular complexes. High-resolution cryo-EM reconstruction often requires hundreds of thousands of single-particle images. Particle extraction from experimental micrographs thus can be laborious and presents a major practical bottleneck in cryo-EM structural determination. Existing computational methods of particle picking often use low-resolution templates as inputs for particle matching, making it possible to cause reference-dependent bias. It is critical to develop a highly efficient template-free method to automatically recognize particle images from cryo-EM micrographs. Results: We developed a deep learning-based algorithmic framework, DeepEM, for single-particle recognition from noisy cryo-EM micrographs, enabling automated particle picking, selection and verification in an integrated fashion. The kernel of DeepEM is built upon a convolutional neural network (CNN) of eight layers, which can be recursively trained to be highly "knowledgeable". Our approach exhibits improved performance and high precision when tested on the standard KLH dataset. Application of DeepEM to several challenging experimental cryo-EM datasets demonstrates its capability in avoiding selection of un-wanted particles and non-particles even when true particles contain fewer features. Conclusions: The DeepEM method derived from a deep CNN allows automated particle extraction from raw cryo-EM micrographs in the absence of templates, which demonstrated improved performance, objectivity and accuracy. Application of this novel approach is expected to free the labor involved in single-particle verification, thus promoting the efficiency of cryo-EM data processing.
* 26 pages, 6 figures, 1 table