 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExploring Temporal Event Cues for Dense Video Captioning in Cyclic Co-learning
Dec 16, 2024



Dense video captioning aims to detect and describe all events in untrimmed videos. This paper presents a dense video captioning network called Multi-Concept Cyclic Learning (MCCL), which aims to: (1) detect multiple concepts at the frame level, using these concepts to enhance video features and provide temporal event cues; and (2) design cyclic co-learning between the generator and the localizer within the captioning network to promote semantic perception and event localization. Specifically, we perform weakly supervised concept detection for each frame, and the detected concept embeddings are integrated into the video features to provide event cues. Additionally, video-level concept contrastive learning is introduced to obtain more discriminative concept embeddings. In the captioning network, we establish a cyclic co-learning strategy where the generator guides the localizer for event localization through semantic matching, while the localizer enhances the generator's event semantic perception through location matching, making semantic perception and event localization mutually beneficial. MCCL achieves state-of-the-art performance on the ActivityNet Captions and YouCook2 datasets. Extensive experiments demonstrate its effectiveness and interpretability.
Prediction of superconducting properties of materials based on machine learning models
Nov 06, 2022



The application of superconducting materials is becoming more and more widespread. Traditionally, the discovery of new superconducting materials relies on the experience of experts and a large number of "trial and error" experiments, which not only increases the cost of experiments but also prolongs the period of discovering new superconducting materials. In recent years, machine learning has been increasingly applied to materials science. Based on this, this manuscript proposes the use of XGBoost model to identify superconductors; the first application of deep forest model to predict the critical temperature of superconductors; the first application of deep forest to predict the band gap of materials; and application of a new sub-network model to predict the Fermi energy level of materials. Compared with our known similar literature, all the above algorithms reach state-of-the-art. Finally, this manuscript uses the above models to search the COD public dataset and identify 50 candidate superconducting materials with possible critical temperature greater than 90 K.
Prediction of properties of metal alloy materials based on machine learning
Sep 20, 2021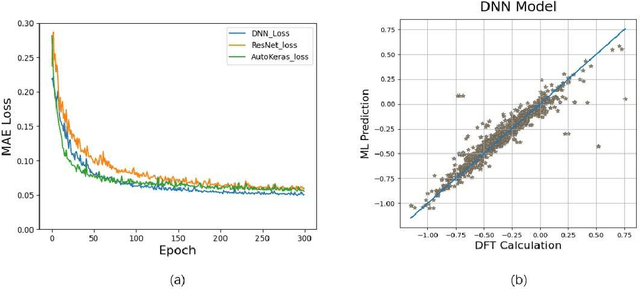
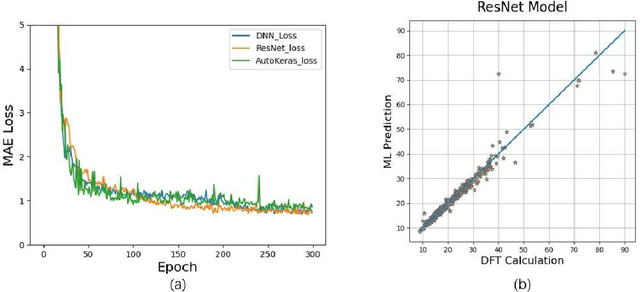
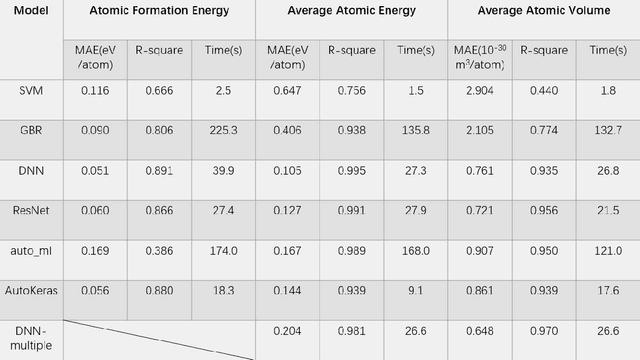
Density functional theory and its optimization algorithm are the main methods to calculate the properties in the field of materials. Although the calculation results are accurate, it costs a lot of time and money. In order to alleviate this problem, we intend to use machine learning to predict material properties. In this paper, we conduct experiments on atomic volume, atomic energy and atomic formation energy of metal alloys, using the open quantum material database. Through the traditional machine learning models, deep learning network and automated machine learning, we verify the feasibility of machine learning in material property prediction. The experimental results show that the machine learning can predict the material properties accurately.
Molecular structure prediction based on graph convolutional networks
Jul 01, 2021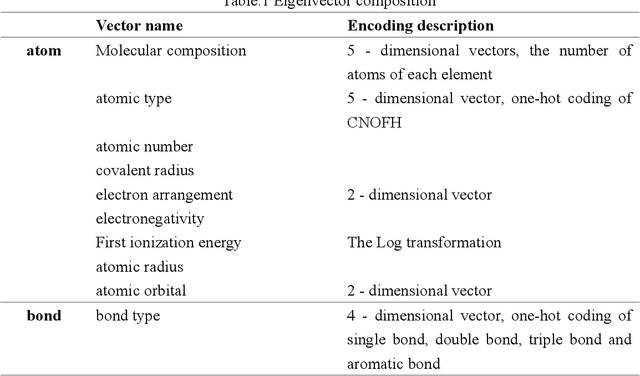
Due to the important application of molecular structure in many fields, calculation by experimental means or traditional density functional theory is often time consuming. In view of this, a new Model Structure based on Graph Convolutional Neural network (MSGCN) is proposed, which can determine the molecular structure by predicting the distance between two atoms. In order to verify the effect of MSGCN model, the model is compared with the method of calculating molecular three-dimensional conformation in RDKit, and the result is better than it. In addition, the distance predicted by the MSGCN model and the distance calculated by the QM9 dataset were used to predict the molecular properties, thus proving the effectiveness of the distance predicted by the MSGCN model.