 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular structure prediction based on graph convolutional networks
Paper and Code
Jul 01, 2021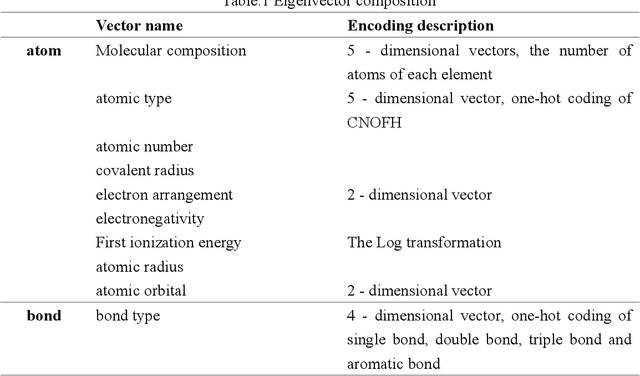
Due to the important application of molecular structure in many fields, calculation by experimental means or traditional density functional theory is often time consuming. In view of this, a new Model Structure based on Graph Convolutional Neural network (MSGCN) is proposed, which can determine the molecular structure by predicting the distance between two atoms. In order to verify the effect of MSGCN model, the model is compared with the method of calculating molecular three-dimensional conformation in RDKit, and the result is better than it. In addition, the distance predicted by the MSGCN model and the distance calculated by the QM9 dataset were used to predict the molecular properties, thus proving the effectiveness of the distance predicted by the MSGCN model.