 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVitaminP: cross-modal learning enables whole-cell segmentation from routine histology
Apr 26, 2026Accurate whole-cell and nuclear segmentation is essential for precision pathology and spatial omics, yet routine hematoxylin and eosin (H&E) staining provides limited cytoplasmic contrast, restricting analyses to nuclei. Multiplex immunofluorescence (mIF) facilitates precise whole-cell delineation but remains constrained by cost and accessibility. We introduce VitaminP, a cross-modal learning framework enabling whole cell segmentation from H&E images. By learning from paired H&E-mIF data, VitaminP transfers molecular boundary information from mIF to overcome cytoplasmic contrast in H&E, establishing cross-modal supervision as a general strategy for recovering missing biological structure. We train VitaminP on 14 public datasets covering 34 cancer types and over 7 million instances, integrating publicly available labels with extensive annotations generated in this study, forming one of the largest resources for segmentation. VitaminP outperforms four state-of-the-art methods and generalizes to unseen datasets, including an in-house dataset spanning 24 rare cancer types. We further developed VitaminPScope, an open-source platform providing an interface for scalable inference and enabling broad adoption.
A deep learning pipeline for PAM50 subtype classification using histopathology images and multi-objective patch selection
Apr 02, 2026Breast cancer is a highly heterogeneous disease with diverse molecular profiles. The PAM50 gene signature is widely recognized as a standard for classifying breast cancer into intrinsic subtypes, enabling more personalized treatment strategies. In this study, we introduce a novel optimization-driven deep learning framework that aims to reduce reliance on costly molecular assays by directly predicting PAM50 subtypes from H&E-stained whole-slide images (WSIs). Our method jointly optimizes patch informativeness, spatial diversity, uncertainty, and patch count by combining the non-dominated sorting genetic algorithm II (NSGA-II) with Monte Carlo dropout-based uncertainty estimation. The proposed method can identify a small but highly informative patch subset for classification. We used a ResNet18 backbone for feature extraction and a custom CNN head for classification. For evaluation, we used the internal TCGA-BRCA dataset as the training cohort and the external CPTAC-BRCA dataset as the test cohort. On the internal dataset, an F1-score of 0.8812 and an AUC of 0.9841 using 627 WSIs from the TCGA-BRCA cohort were achieved. The performance of the proposed approach on the external validation dataset showed an F1-score of 0.7952 and an AUC of 0.9512. These findings indicate that the proposed optimization-guided, uncertainty-aware patch selection can achieve high performance and improve the computational efficiency of histopathology-based PAM50 classification compared to existing methods, suggesting a scalable imaging-based replacement that has the potential to support clinical decision-making.
Prototype-driven fusion of pathology and spatial transcriptomics for interpretable survival prediction
Feb 12, 2026Whole slide images (WSIs) enable weakly supervised prognostic modeling via multiple instance learning (MIL). Spatial transcriptomics (ST) preserves in situ gene expression, providing a spatial molecular context that complements morphology. As paired WSI-ST cohorts scale to population level, leveraging their complementary spatial signals for prognosis becomes crucial; however, principled cross-modal fusion strategies remain limited for this paradigm. To this end, we introduce PathoSpatial, an interpretable end-to-end framework integrating co-registered WSIs and ST to learn spatially informed prognostic representations. PathoSpatial uses task-guided prototype learning within a multi-level experts architecture, adaptively orchestrating unsupervised within-modality discovery with supervised cross-modal aggregation. By design, PathoSpatial substantially strengthens interpretability while maintaining discriminative ability. We evaluate PathoSpatial on a triple-negative breast cancer cohort with paired ST and WSIs. PathoSpatial delivers strong and consistent performance across five survival endpoints, achieving superior or comparable performance to leading unimodal and multimodal methods. PathoSpatial inherently enables post-hoc prototype interpretation and molecular risk decomposition, providing quantitative, biologically grounded explanations, highlighting candidate prognostic factors. We present PathoSpatial as a proof-of-concept for scalable and interpretable multimodal learning for spatial omics-pathology fusion.
CellSymphony: Deciphering the molecular and phenotypic orchestration of cells with single-cell pathomics
Aug 13, 2025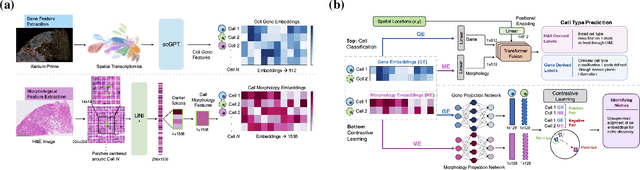
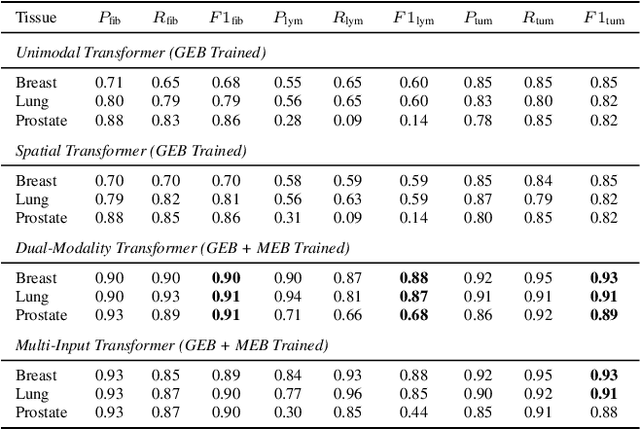
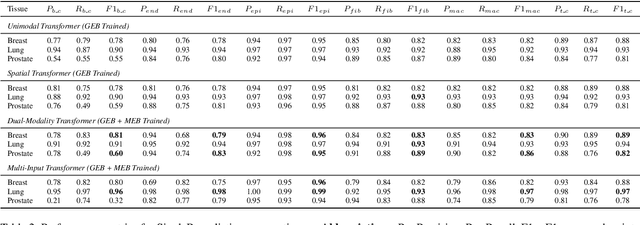
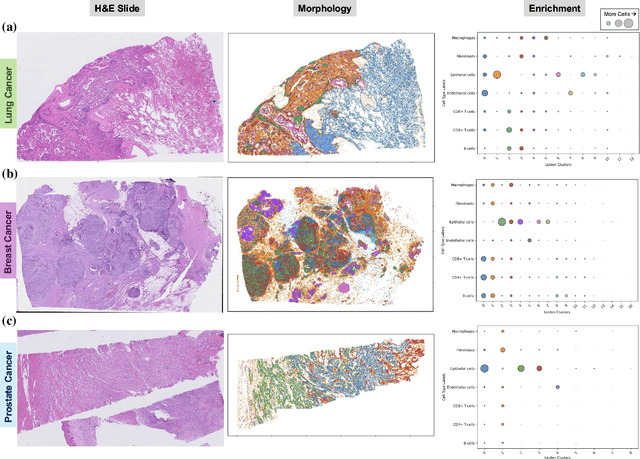
Xenium, a new spatial transcriptomics platform, enables subcellular-resolution profiling of complex tumor tissues. Despite the rich morphological information in histology images, extracting robust cell-level features and integrating them with spatial transcriptomics data remains a critical challenge. We introduce CellSymphony, a flexible multimodal framework that leverages foundation model-derived embeddings from both Xenium transcriptomic profiles and histology images at true single-cell resolution. By learning joint representations that fuse spatial gene expression with morphological context, CellSymphony achieves accurate cell type annotation and uncovers distinct microenvironmental niches across three cancer types. This work highlights the potential of foundation models and multimodal fusion for deciphering the physiological and phenotypic orchestration of cells within complex tissue ecosystems.
Cell abundance aware deep learning for cell detection on highly imbalanced pathological data
Feb 23, 2021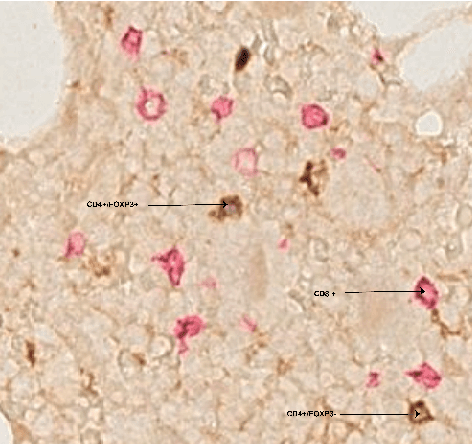


Automated analysis of tissue sections allows a better understanding of disease biology and may reveal biomarkers that could guide prognosis or treatment selection. In digital pathology, less abundant cell types can be of biological significance, but their scarcity can result in biased and sub-optimal cell detection model. To minimize the effect of cell imbalance on cell detection, we proposed a deep learning pipeline that considers the abundance of cell types during model training. Cell weight images were generated, which assign larger weights to less abundant cells and used the weights to regularize Dice overlap loss function. The model was trained and evaluated on myeloma bone marrow trephine samples. Our model obtained a cell detection F1-score of 0.78, a 2% increase compared to baseline models, and it outperformed baseline models at detecting rare cell types. We found that scaling deep learning loss function by the abundance of cells improves cell detection performance. Our results demonstrate the importance of incorporating domain knowledge on deep learning methods for pathological data with class imbalance.
Glioma Classification Using Multimodal Radiology and Histology Data
Nov 10, 2020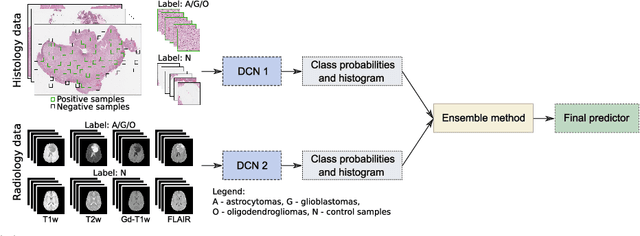
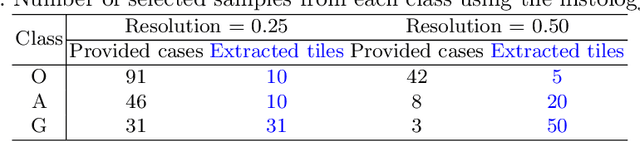
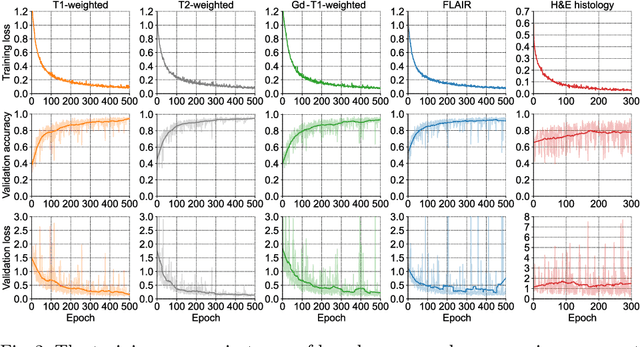
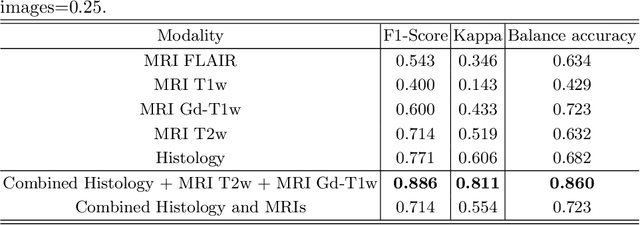
Gliomas are brain tumours with a high mortality rate. There are various grades and sub-types of this tumour, and the treatment procedure varies accordingly. Clinicians and oncologists diagnose and categorise these tumours based on visual inspection of radiology and histology data. However, this process can be time-consuming and subjective. The computer-assisted methods can help clinicians to make better and faster decisions. In this paper, we propose a pipeline for automatic classification of gliomas into three sub-types: oligodendroglioma, astrocytoma, and glioblastoma, using both radiology and histopathology images. The proposed approach implements distinct classification models for radiographic and histologic modalities and combines them through an ensemble method. The classification algorithm initially carries out tile-level (for histology) and slice-level (for radiology) classification via a deep learning method, then tile/slice-level latent features are combined for a whole-slide and whole-volume sub-type prediction. The classification algorithm was evaluated using the data set provided in the CPM-RadPath 2020 challenge. The proposed pipeline achieved the F1-Score of 0.886, Cohen's Kappa score of 0.811 and Balance accuracy of 0.860. The ability of the proposed model for end-to-end learning of diverse features enables it to give a comparable prediction of glioma tumour sub-types.
ConCORDe-Net: Cell Count Regularized Convolutional Neural Network for Cell Detection in Multiplex Immunohistochemistry Images
Aug 01, 2019
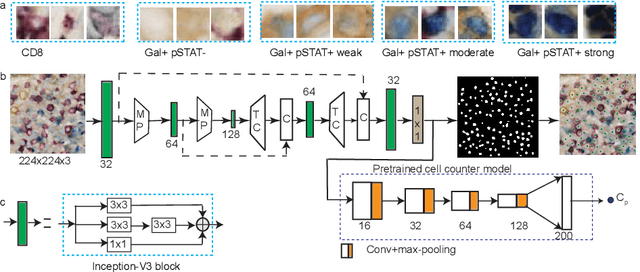

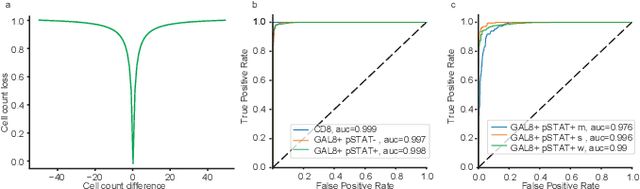
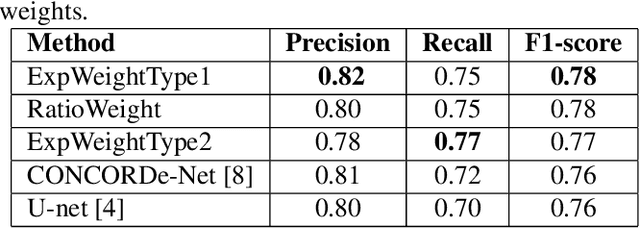
In digital pathology, cell detection and classification are often prerequisites to quantify cell abundance and explore tissue spatial heterogeneity. However, these tasks are particularly challenging for multiplex immunohistochemistry (mIHC) images due to high levels of variability in staining, expression intensity, and inherent noise as a result of preprocessing artefacts. We proposed a deep learning method to detect and classify cells in mIHC whole-tumour slide images of breast cancer. Inspired by inception-v3, we developed Cell COunt RegularizeD Convolutional neural Network (ConCORDe-Net) which integrates conventional dice overlap and a new cell count loss function for optimizing cell detection, followed by a multi-stage convolutional neural network for cell classification. In total, 20447 cells, belonging to five cell classes were annotated by experts from 175 patches extracted from 6 whole-tumour mIHC images. These patches were randomly split into training, validation and testing sets. Using ConCORDe-Net, we obtained a cell detection F1 score of 0.873, which is the best score compared to three state of the art methods. In particular, ConCORDe-Net excels at detecting closely located and weakly stained cells compared to other methods. Incorporating cell count loss in the objective function regularizes the network to learn weak gradient boundaries and separate weakly stained cells from background artefacts. Moreover, cell classification accuracy of 96.5% was achieved. These results support that incorporating problem-specific knowledge such as cell count into deep learning-based cell detection architectures improve the robustness of the algorithm.
Capturing global spatial context for accurate cell classification in skin cancer histology
Aug 07, 2018



The spectacular response observed in clinical trials of immunotherapy in patients with previously uncurable Melanoma, a highly aggressive form of skin cancer, calls for a better understanding of the cancer-immune interface. Computational pathology provides a unique opportunity to spatially dissect such interface on digitised pathological slides. Accurate cellular classification is a key to ensure meaningful results, but is often challenging even with state-of-art machine learning and deep learning methods. We propose a hierarchical framework, which mirrors the way pathologists perceive tumour architecture and define tumour heterogeneity to improve cell classification methods that rely solely on cell nuclei morphology. The SLIC superpixel algorithm was used to segment and classify tumour regions in low resolution H&E-stained histological images of melanoma skin cancer to provide a global context. Classification of superpixels into tumour, stroma, epidermis and lumen/white space, yielded a 97.7% training set accuracy and 95.7% testing set accuracy in 58 whole-tumour images of the TCGA melanoma dataset. The superpixel classification was projected down to high resolution images to enhance the performance of a single cell classifier, based on cell nuclear morphological features, and resulted in increasing its accuracy from 86.4% to 91.6%. Furthermore, a voting scheme was proposed to use global context as biological a priori knowledge, pushing the accuracy further to 92.8%. This study demonstrates how using the global spatial context can accurately characterise the tumour microenvironment and allow us to extend significantly beyond single-cell morphological classification.
DeepSDCS: Dissecting cancer proliferation heterogeneity in Ki67 digital whole slide images
Jun 28, 2018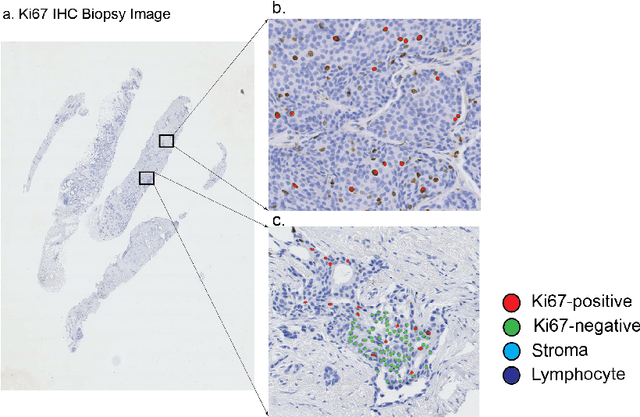

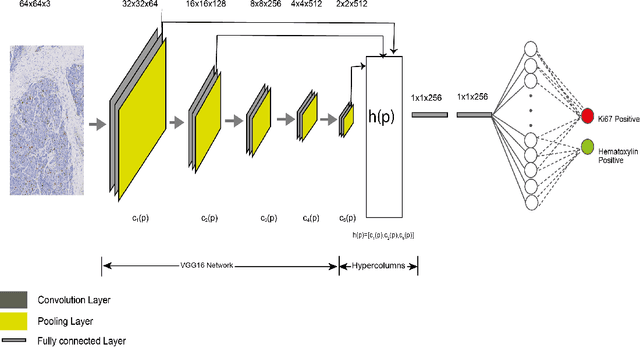

Ki67 is an important biomarker for breast cancer. Classification of positive and negative Ki67 cells in histology slides is a common approach to determine cancer proliferation status. However, there is a lack of generalizable and accurate methods to automate Ki67 scoring in large-scale patient cohorts. In this work, we have employed a novel deep learning technique based on hypercolumn descriptors for cell classification in Ki67 images. Specifically, we developed the Simultaneous Detection and Cell Segmentation (DeepSDCS) network to perform cell segmentation and detection. VGG16 network was used for the training and fine tuning to training data. We extracted the hypercolumn descriptors of each cell to form the vector of activation from specific layers to capture features at different granularity. Features from these layers that correspond to the same pixel were propagated using a stochastic gradient descent optimizer to yield the detection of the nuclei and the final cell segmentations. Subsequently, seeds generated from cell segmentation were propagated to a spatially constrained convolutional neural network for the classification of the cells into stromal, lymphocyte, Ki67-positive cancer cell, and Ki67-negative cancer cell. We validated its accuracy in the context of a large-scale clinical trial of oestrogen-receptor-positive breast cancer. We achieved 99.06% and 89.59% accuracy on two separate test sets of Ki67 stained breast cancer dataset comprising biopsy and whole-slide images.
Deconvolving convolution neural network for cell detection
Jun 18, 2018
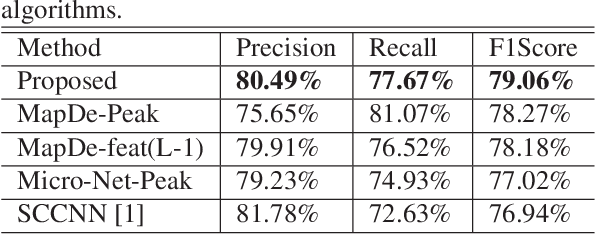
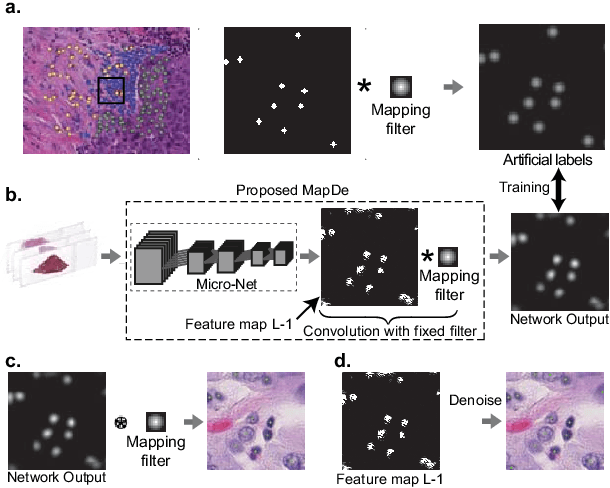
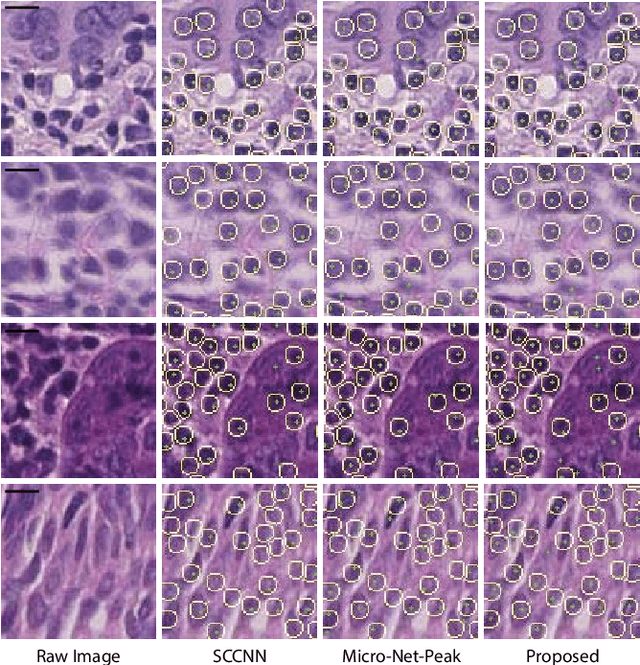
Automatic cell detection in histology images is a challenging task due to varying size, shape and features of cells and stain variations across a large cohort. Conventional deep learning methods regress the probability of each pixel belonging to the centre of a cell followed by detection of local maxima. We present deconvolution as an alternate approach to local maxima detection. The ground truth points are convolved with a mapping filter to generate artifical labels. A convolutional neural network (CNN) is modified to convolve it's output with the same mapping filter and is trained for the mapped labels. Output of the trained CNN is then deconvolved to generate points as cell detection. We compare our method with state-of-the-art deep learning approaches where the results show that the proposed approach detects cells with comparatively high precision and F1-score.