 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStructure-aware Interactive Graph Neural Networks for the Prediction of Protein-Ligand Binding Affinity
Jul 21, 2021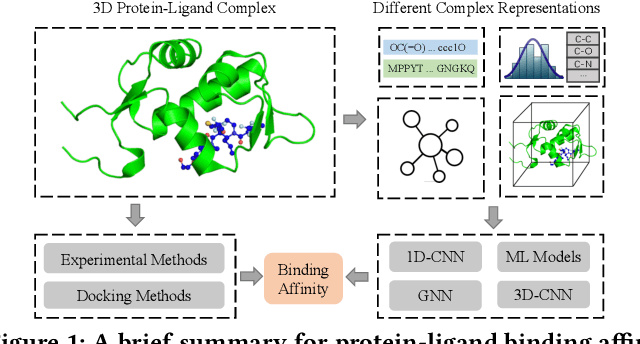
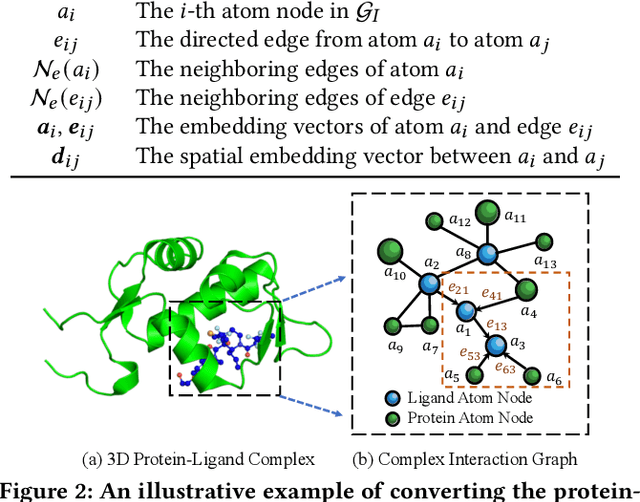
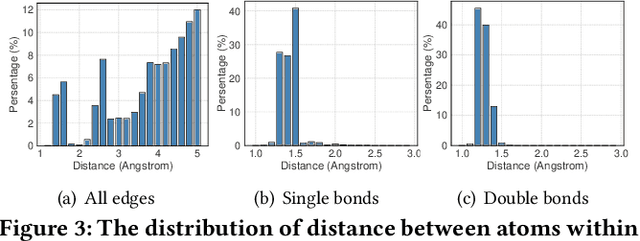
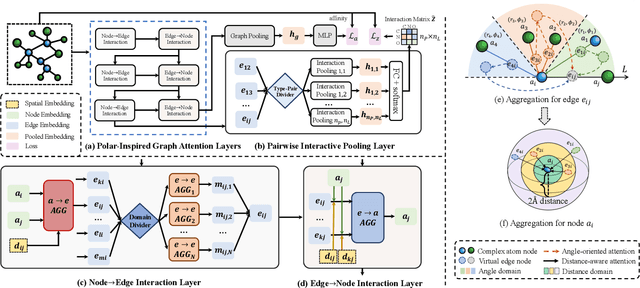
Drug discovery often relies on the successful prediction of protein-ligand binding affinity. Recent advances have shown great promise in applying graph neural networks (GNNs) for better affinity prediction by learning the representations of protein-ligand complexes. However, existing solutions usually treat protein-ligand complexes as topological graph data, thus the biomolecular structural information is not fully utilized. The essential long-range interactions among atoms are also neglected in GNN models. To this end, we propose a structure-aware interactive graph neural network (SIGN) which consists of two components: polar-inspired graph attention layers (PGAL) and pairwise interactive pooling (PiPool). Specifically, PGAL iteratively performs the node-edge aggregation process to update embeddings of nodes and edges while preserving the distance and angle information among atoms. Then, PiPool is adopted to gather interactive edges with a subsequent reconstruction loss to reflect the global interactions. Exhaustive experimental study on two benchmarks verifies the superiority of SIGN.