 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRe-analysis of the Human Transcription Factor Atlas Recovers TF-Specific Signatures from Pooled Single-Cell Screens with Missing Controls
Apr 02, 2026Public pooled single-cell perturbation atlases are valuable resources for studying transcription factor (TF) function, but downstream re-analysis can be limited by incomplete deposited metadata and missing internal controls. Here we re-analyze the human TF Atlas dataset (GSE216481), a MORF-based pooled overexpression screen spanning 3,550 TF open reading frames and 254,519 cells, with a reproducible pipeline for quality control, MORF barcode demultiplexing, per-TF differential expression, and functional enrichment. From 77,018 cells in the pooled screen, we assign 60,997 (79.2\%) to 87 TF identities. Because the deposited barcode mapping lacks the GFP and mCherry negative controls present in the original library, we use embryoid body (EB) cells as an external baseline and remove shared batch/transduction artifacts by background subtraction. This strategy recovers TF-specific signatures for 59 of 61 testable TFs, compared with 27 detected by one-vs-rest alone, showing that robust TF-level signal can be rescued despite missing intra-pool controls. HOPX, MAZ, PAX6, FOS, and FEZF2 emerge as the strongest transcriptional remodelers, while per-TF enrichment links FEZF2 to regulation of differentiation, EGR1 to Hippo and cardiac programs, FOS to focal adhesion, and NFIC to collagen biosynthesis. Condition-level analyses reveal convergent Wnt, neurogenic, EMT, and Hippo signatures, and Harmony indicates minimal confounding batch effects across pooled replicates. Our per-TF effect sizes significantly agree with Joung et al.'s published rankings (Spearman $ρ= -0.316$, $p = 0.013$; negative because lower rank indicates stronger effect). Together, these results show that the deposited TF Atlas data can support validated TF-specific transcriptional and pathway analyses when paired with principled external controls, artifact removal, and reproducible computation.
Multimodal spatiotemporal graph neural networks for improved prediction of 30-day all-cause hospital readmission
Apr 14, 2022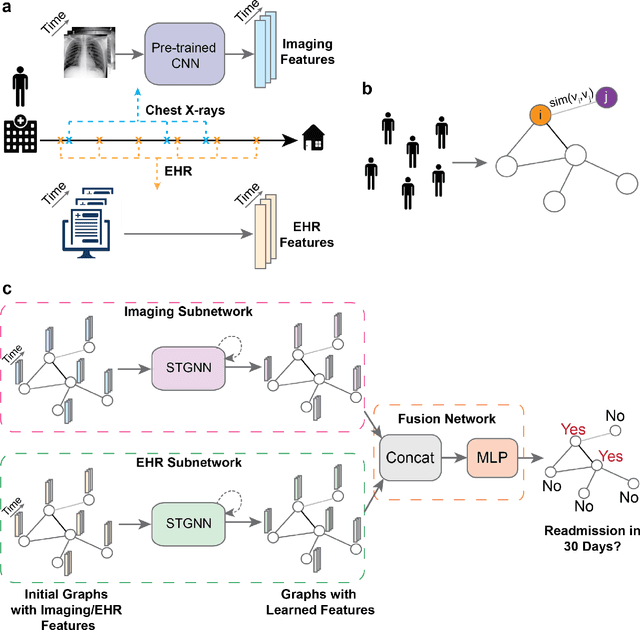
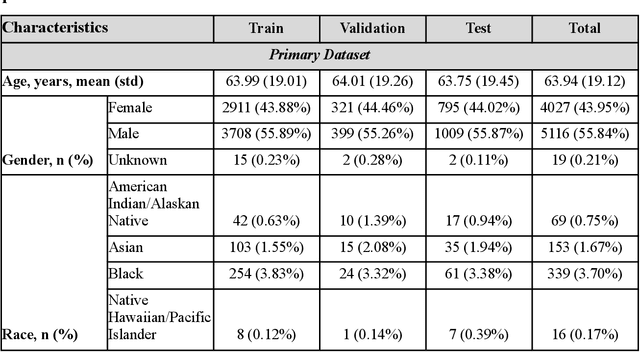
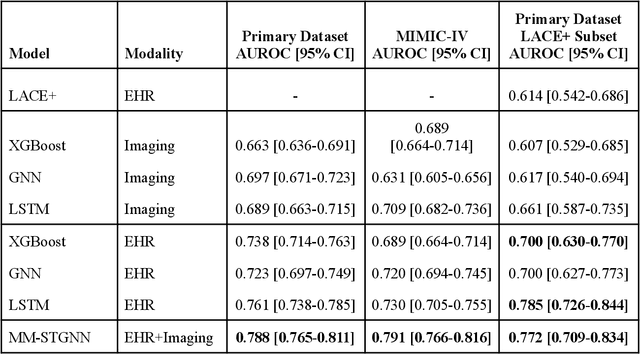
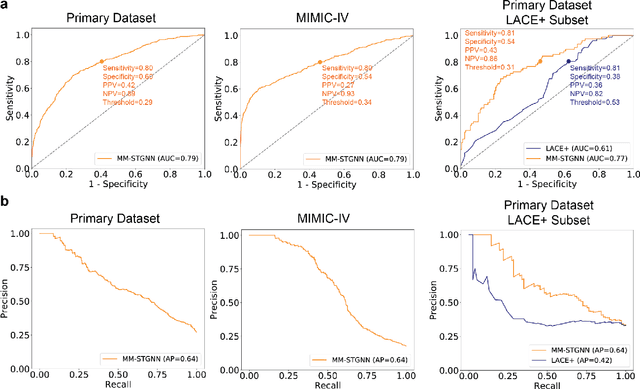
Measures to predict 30-day readmission are considered an important quality factor for hospitals as accurate predictions can reduce the overall cost of care by identifying high risk patients before they are discharged. While recent deep learning-based studies have shown promising empirical results on readmission prediction, several limitations exist that may hinder widespread clinical utility, such as (a) only patients with certain conditions are considered, (b) existing approaches do not leverage data temporality, (c) individual admissions are assumed independent of each other, which is unrealistic, (d) prior studies are usually limited to single source of data and single center data. To address these limitations, we propose a multimodal, modality-agnostic spatiotemporal graph neural network (MM-STGNN) for prediction of 30-day all-cause hospital readmission that fuses multimodal in-patient longitudinal data. By training and evaluating our methods using longitudinal chest radiographs and electronic health records from two independent centers, we demonstrate that MM-STGNN achieves AUROC of 0.79 on both primary and external datasets. Furthermore, MM-STGNN significantly outperforms the current clinical reference standard, LACE+ score (AUROC=0.61), on the primary dataset. For subset populations of patients with heart and vascular disease, our model also outperforms baselines on predicting 30-day readmission (e.g., 3.7 point improvement in AUROC in patients with heart disease). Lastly, qualitative model interpretability analysis indicates that while patients' primary diagnoses were not explicitly used to train the model, node features crucial for model prediction directly reflect patients' primary diagnoses. Importantly, our MM-STGNN is agnostic to node feature modalities and could be utilized to integrate multimodal data for triaging patients in various downstream resource allocation tasks.