 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUncertainty Quantification in Graph Neural Networks with Shallow Ensembles
Apr 17, 2025Machine-learned potentials (MLPs) have revolutionized materials discovery by providing accurate and efficient predictions of molecular and material properties. Graph Neural Networks (GNNs) have emerged as a state-of-the-art approach due to their ability to capture complex atomic interactions. However, GNNs often produce unreliable predictions when encountering out-of-domain data and it is difficult to identify when that happens. To address this challenge, we explore Uncertainty Quantification (UQ) techniques, focusing on Direct Propagation of Shallow Ensembles (DPOSE) as a computationally efficient alternative to deep ensembles. By integrating DPOSE into the SchNet model, we assess its ability to provide reliable uncertainty estimates across diverse Density Functional Theory datasets, including QM9, OC20, and Gold Molecular Dynamics. Our findings often demonstrate that DPOSE successfully distinguishes between in-domain and out-of-domain samples, exhibiting higher uncertainty for unobserved molecule and material classes. This work highlights the potential of lightweight UQ methods in improving the robustness of GNN-based materials modeling and lays the foundation for future integration with active learning strategies.
Reflections from the 2024 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
Nov 20, 2024
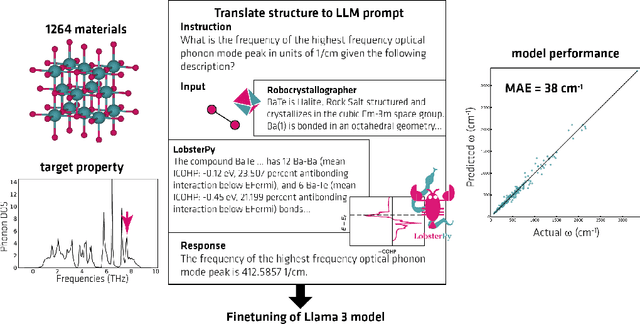
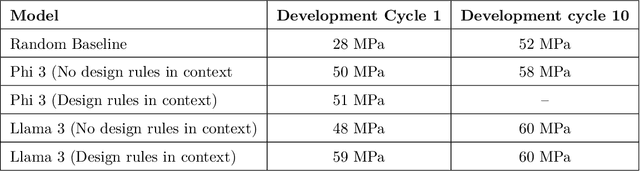
Here, we present the outcomes from the second Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry, which engaged participants across global hybrid locations, resulting in 34 team submissions. The submissions spanned seven key application areas and demonstrated the diverse utility of LLMs for applications in (1) molecular and material property prediction; (2) molecular and material design; (3) automation and novel interfaces; (4) scientific communication and education; (5) research data management and automation; (6) hypothesis generation and evaluation; and (7) knowledge extraction and reasoning from scientific literature. Each team submission is presented in a summary table with links to the code and as brief papers in the appendix. Beyond team results, we discuss the hackathon event and its hybrid format, which included physical hubs in Toronto, Montreal, San Francisco, Berlin, Lausanne, and Tokyo, alongside a global online hub to enable local and virtual collaboration. Overall, the event highlighted significant improvements in LLM capabilities since the previous year's hackathon, suggesting continued expansion of LLMs for applications in materials science and chemistry research. These outcomes demonstrate the dual utility of LLMs as both multipurpose models for diverse machine learning tasks and platforms for rapid prototyping custom applications in scientific research.
Adsorb-Agent: Autonomous Identification of Stable Adsorption Configurations via Large Language Model Agent
Oct 22, 2024Adsorption energy is a key reactivity descriptor in catalysis, enabling the efficient screening of potential catalysts. However, determining adsorption energy involves comparing the energies of multiple adsorbate-catalyst configurations, which is computationally demanding due to a large number of possible configurations. Current algorithmic approaches typically enumerate adsorption sites and configurations without leveraging theoretical insights to guide the initial setup. In this work, we present Adsorb-Agent, a Large Language Model (LLM) agent designed to efficiently derive system-specific stable adsorption configurations with minimal human intervention. Adsorb-Agent leverages built-in knowledge and emergent reasoning capabilities, significantly reducing the number of initial configurations required while improving accuracy in predicting the minimum adsorption energy. We demonstrate its performance using two example systems, NNH-CuPd3 (111) and NNH-Mo3Pd (111), for the Nitrogen Reduction Reaction (NRR), a sustainable alternative to the Haber-Bosch process. Adsorb-Agent outperforms conventional "heuristic" and "random" algorithms by identifying lower-energy configurations with fewer initial setups, reducing computational costs while enhancing accuracy. This highlights its potential to accelerate catalyst discovery.
Explainable Data-driven Modeling of Adsorption Energy in Heterogeneous Catalysis
May 30, 2024The increasing popularity of machine learning (ML) in catalysis has spurred interest in leveraging these techniques to enhance catalyst design. Our study aims to bridge the gap between physics-based studies and data-driven methodologies by integrating ML techniques with eXplainable AI (XAI). Specifically, we employ two XAI techniques: Post-hoc XAI analysis and Symbolic Regression. These techniques help us unravel the correlation between adsorption energy and the properties of the adsorbate-catalyst system. Leveraging a large dataset such as the Open Catalyst Dataset (OC20), we employ a combination of shallow ML techniques and XAI methodologies. Our investigation involves utilizing multiple shallow machine learning techniques to predict adsorption energy, followed by post-hoc analysis for feature importance, inter-feature correlations, and the influence of various feature values on the prediction of adsorption energy. The post-hoc analysis reveals that adsorbate properties exert a greater influence than catalyst properties in our dataset. The top five features based on higher Shapley values are adsorbate electronegativity, the number of adsorbate atoms, catalyst electronegativity, effective coordination number, and the sum of atomic numbers of the adsorbate molecule. There is a positive correlation between catalyst and adsorbate electronegativity with the prediction of adsorption energy. Additionally, symbolic regression yields results consistent with SHAP analysis. It deduces a mathematical relationship indicating that the square of the catalyst electronegativity is directly proportional to the adsorption energy. These consistent correlations resemble those derived from physics-based equations in previous research. Our work establishes a robust framework that integrates ML techniques with XAI, leveraging large datasets like OC20 to enhance catalyst design through model explainability.