 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge3-D Representations for Hyperspectral Flame Tomography
Mar 29, 2026Flame tomography is a compelling approach for extracting large amounts of data from experiments via 3-D thermochemical reconstruction. Recent efforts employing neural-network flame representations have suggested improved reconstruction quality compared with classical tomography approaches, but a rigorous quantitative comparison with the same algorithm using a voxel-grid representation has not been conducted. Here, we compare a classical voxel-grid representation with varying regularizers to a continuous neural representation for tomographic reconstruction of a simulated pool fire. The representations are constructed to give temperature and composition as a function of location, and a subsequent ray-tracing step is used to solve the radiative transfer equation to determine the spectral intensity incident on hyperspectral infrared cameras, which is then convolved with an instrument lineshape function. We demonstrate that the voxel-grid approach with a total-variation regularizer reproduces the ground-truth synthetic flame with the highest accuracy for reduced memory intensity and runtime. Future work will explore more representations and under experimental configurations.
GLU: Global-Local-Uncertainty Fusion for Scalable Spatiotemporal Reconstruction and Forecasting
Mar 27, 2026Digital twins of complex physical systems are expected to infer unobserved states from sparse measurements and predict their evolution in time, yet these two functions are typically treated as separate tasks. Here we present GLU, a Global-Local-Uncertainty framework that formulates sparse reconstruction and dynamic forecasting as a unified state-representation problem and introduces a structured latent assembly to both tasks. The central idea is to build a structured latent state that combines a global summary of system-level organization, local tokens anchored to available measurements, and an uncertainty-driven importance field that weights observations according to the physical informativeness. For reconstruction, GLU uses importance-aware adaptive neighborhood selection to retrieve locally relevant information while preserving global consistency and allowing flexible query resolution on arbitrary geometries. Across a suite of challenging benchmarks, GLU consistently improves reconstruction fidelity over reduced-order, convolutional, neural operator, and attention-based baselines, better preserving multi-scale structures. For forecasting, a hierarchical Leader-Follower Dynamics module evolves the latent state with substantially reduced memory growth, maintains stable rollout behavior and delays error accumulation in nonlinear dynamics. On a realistic turbulent combustion dataset, it further preserves not only sharp fronts and broadband structures in multiple physical fields, but also their cross-channel thermo-chemical couplings. Scalability tests show that these gains are achieved with substantially lower memory growth than comparable attention-based baselines. Together, these results establish GLU as a flexible and computationally practical paradigm for sparse digital twins.
Latent Generative Solvers for Generalizable Long-Term Physics Simulation
Feb 11, 2026We study long-horizon surrogate simulation across heterogeneous PDE systems. We introduce Latent Generative Solvers (LGS), a two-stage framework that (i) maps diverse PDE states into a shared latent physics space with a pretrained VAE, and (ii) learns probabilistic latent dynamics with a Transformer trained by flow matching. Our key mechanism is an uncertainty knob that perturbs latent inputs during training and inference, teaching the solver to correct off-manifold rollout drift and stabilizing autoregressive prediction. We further use flow forcing to update a system descriptor (context) from model-generated trajectories, aligning train/test conditioning and improving long-term stability. We pretrain on a curated corpus of $\sim$2.5M trajectories at $128^2$ resolution spanning 12 PDE families. LGS matches strong deterministic neural-operator baselines on short horizons while substantially reducing rollout drift on long horizons. Learning in latent space plus efficient architectural choices yields up to \textbf{70$\times$} lower FLOPs than non-generative baselines, enabling scalable pretraining. We also show efficient adaptation to an out-of-distribution $256^2$ Kolmogorov flow dataset under limited finetuning budgets. Overall, LGS provides a practical route toward generalizable, uncertainty-aware neural PDE solvers that are more reliable for long-term forecasting and downstream scientific workflows.
Learning continuous SOC-dependent thermal decomposition kinetics for Li-ion cathodes using KA-CRNNs
Dec 17, 2025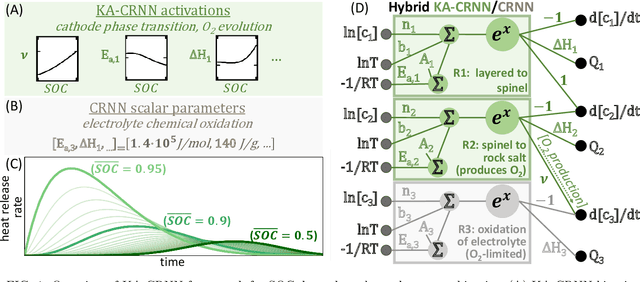
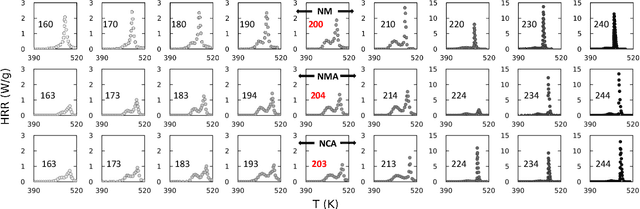
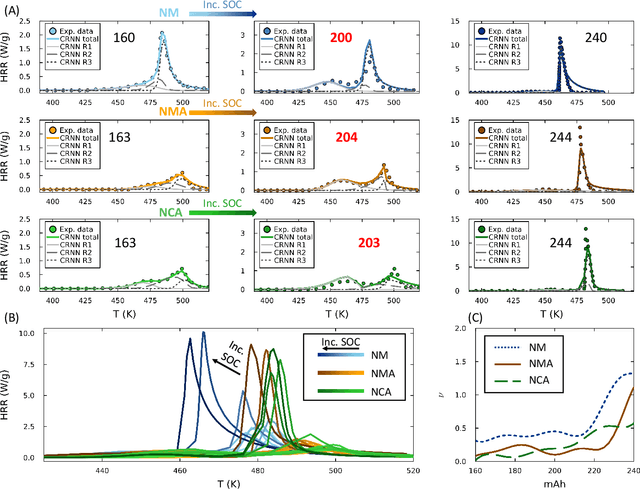
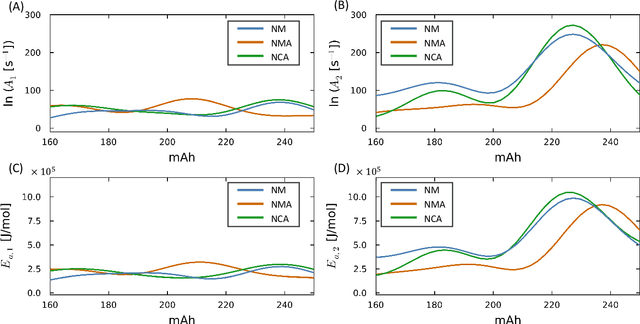
Thermal runaway in lithium-ion batteries is strongly influenced by the state of charge (SOC). Existing predictive models typically infer scalar kinetic parameters at a full SOC or a few discrete SOC levels, preventing them from capturing the continuous SOC dependence that governs exothermic behavior during abuse conditions. To address this, we apply the Kolmogorov-Arnold Chemical Reaction Neural Network (KA-CRNN) framework to learn continuous and realistic SOC-dependent exothermic cathode-electrolyte interactions. We apply a physics-encoded KA-CRNN to learn SOC-dependent kinetic parameters for cathode-electrolyte decomposition directly from differential scanning calorimetry (DSC) data. A mechanistically informed reaction pathway is embedded into the network architecture, enabling the activation energies, pre-exponential factors, enthalpies, and related parameters to be represented as continuous and fully interpretable functions of the SOC. The framework is demonstrated for NCA, NM, and NMA cathodes, yielding models that reproduce DSC heat-release features across all SOCs and provide interpretable insight into SOC-dependent oxygen-release and phase-transformation mechanisms. This approach establishes a foundation for extending kinetic parameter dependencies to additional environmental and electrochemical variables, supporting more accurate and interpretable thermal-runaway prediction and monitoring.
Kolmogorov-Arnold Chemical Reaction Neural Networks for learning pressure-dependent kinetic rate laws
Nov 10, 2025Chemical Reaction Neural Networks (CRNNs) have emerged as an interpretable machine learning framework for discovering reaction kinetics directly from data, while strictly adhering to the Arrhenius and mass action laws. However, standard CRNNs cannot represent pressure-dependent rate behavior, which is critical in many combustion and chemical systems and typically requires empirical formulations such as Troe or PLOG. Here, we develop Kolmogorov-Arnold Chemical Reaction Neural Networks (KA-CRNNs) that generalize CRNNs by modeling each kinetic parameter as a learnable function of system pressure using Kolmogorov-Arnold activations. This structure maintains full interpretability and physical consistency while enabling assumption-free inference of pressure effects directly from data. A proof-of-concept study on the CH3 recombination reaction demonstrates that KA-CRNNs accurately reproduce pressure-dependent kinetics across a range of temperatures and pressures, outperforming conventional interpolative models. The framework establishes a foundation for data-driven discovery of extended kinetic behaviors in complex reacting systems, advancing interpretable and physics-consistent approaches for chemical model inference.
ChemKANs for Combustion Chemistry Modeling and Acceleration
Apr 17, 2025



Efficient chemical kinetic model inference and application for combustion problems is challenging due to large ODE systems and wideley separated time scales. Machine learning techniques have been proposed to streamline these models, though strong nonlinearity and numerical stiffness combined with noisy data sources makes their application challenging. The recently developed Kolmogorov-Arnold Networks (KANs) and KAN ordinary differential equations (KAN-ODEs) have been demonstrated as powerful tools for scientific applications thanks to their rapid neural scaling, improved interpretability, and smooth activation functions. Here, we develop ChemKANs by augmenting the KAN-ODE framework with physical knowledge of the flow of information through the relevant kinetic and thermodynamic laws, as well as an elemental conservation loss term. This novel framework encodes strong inductive bias that enables streamlined training and higher accuracy predictions, while facilitating parameter sparsity through full sharing of information across all inputs and outputs. In a model inference investigation, we find that ChemKANs exhibit no overfitting or model degradation when tasked with extracting predictive models from data that is both sparse and noisy, a task that a standard DeepONet struggles to accomplish. Next, we find that a remarkably parameter-lean ChemKAN (only 344 parameters) can accurately represent hydrogen combustion chemistry, providing a 2x acceleration over the detailed chemistry in a solver that is generalizable to larger-scale turbulent flow simulations. These demonstrations indicate potential for ChemKANs in combustion physics and chemical kinetics, and demonstrate the scalability of generic KAN-ODEs in significantly larger and more numerically challenging problems than previously studied.
LeanKAN: A Parameter-Lean Kolmogorov-Arnold Network Layer with Improved Memory Efficiency and Convergence Behavior
Feb 25, 2025



The recently proposed Kolmogorov-Arnold network (KAN) is a promising alternative to multi-layer perceptrons (MLPs) for data-driven modeling. While original KAN layers were only capable of representing the addition operator, the recently-proposed MultKAN layer combines addition and multiplication subnodes in an effort to improve representation performance. Here, we find that MultKAN layers suffer from a few key drawbacks including limited applicability in output layers, bulky parameterizations with extraneous activations, and the inclusion of complex hyperparameters. To address these issues, we propose LeanKANs, a direct and modular replacement for MultKAN and traditional AddKAN layers. LeanKANs address these three drawbacks of MultKAN through general applicability as output layers, significantly reduced parameter counts for a given network structure, and a smaller set of hyperparameters. As a one-to-one layer replacement for standard AddKAN and MultKAN layers, LeanKAN is able to provide these benefits to traditional KAN learning problems as well as augmented KAN structures in which it serves as the backbone, such as KAN Ordinary Differential Equations (KAN-ODEs) or Deep Operator KANs (DeepOKAN). We demonstrate LeanKAN's simplicity and efficiency in a series of demonstrations carried out across both a standard KAN toy problem and a KAN-ODE dynamical system modeling problem, where we find that its sparser parameterization and compact structure serve to increase its expressivity and learning capability, leading it to outperform similar and even much larger MultKANs in various tasks.
KAN-ODEs: Kolmogorov-Arnold Network Ordinary Differential Equations for Learning Dynamical Systems and Hidden Physics
Jul 05, 2024



Kolmogorov-Arnold Networks (KANs) as an alternative to Multi-layer perceptrons (MLPs) are a recent development demonstrating strong potential for data-driven modeling. This work applies KANs as the backbone of a Neural Ordinary Differential Equation framework, generalizing their use to the time-dependent and grid-sensitive cases often seen in scientific machine learning applications. The proposed KAN-ODEs retain the flexible dynamical system modeling framework of Neural ODEs while leveraging the many benefits of KANs, including faster neural scaling, stronger interpretability, and lower parameter counts when compared against MLPs. We demonstrate these benefits in three test cases: the Lotka-Volterra predator-prey model, Burgers' equation, and the Fisher-KPP PDE. We showcase the strong performance of parameter-lean KAN-ODE systems generally in reconstructing entire dynamical systems, and also in targeted applications to the inference of a source term in an otherwise known flow field. We additionally demonstrate the interpretability of KAN-ODEs via activation function visualization and symbolic regression of trained results. The successful training of KAN-ODEs and their improved performance when compared to traditional Neural ODEs implies significant potential in leveraging this novel network architecture in myriad scientific machine learning applications.
Arrhenius.jl: A Differentiable Combustion SimulationPackage
Jun 19, 2021
Combustion kinetic modeling is an integral part of combustion simulation, and extensive studies have been devoted to developing both high fidelity and computationally affordable models. Despite these efforts, modeling combustion kinetics is still challenging due to the demand for expert knowledge and optimization against experiments, as well as the lack of understanding of the associated uncertainties. Therefore, data-driven approaches that enable efficient discovery and calibration of kinetic models have received much attention in recent years, the core of which is the optimization based on big data. Differentiable programming is a promising approach for learning kinetic models from data by efficiently computing the gradient of objective functions to model parameters. However, it is often challenging to implement differentiable programming in practice. Therefore, it is still not available in widely utilized combustion simulation packages such as CHEMKIN and Cantera. Here, we present a differentiable combustion simulation package leveraging the eco-system in Julia, including DifferentialEquations.jl for solving differential equations, ForwardDiff.jl for auto-differentiation, and Flux.jl for incorporating neural network models into combustion simulations and optimizing neural network models using the state-of-the-art deep learning optimizers. We demonstrate the benefits of differentiable programming in efficient and accurate gradient computations, with applications in uncertainty quantification, kinetic model reduction, data assimilation, and model discovery.
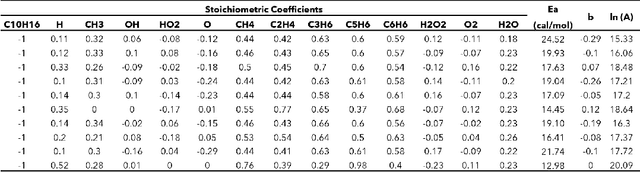
Autonomous Kinetic Modeling of Biomass Pyrolysis using Chemical Reaction Neural Networks
May 24, 2021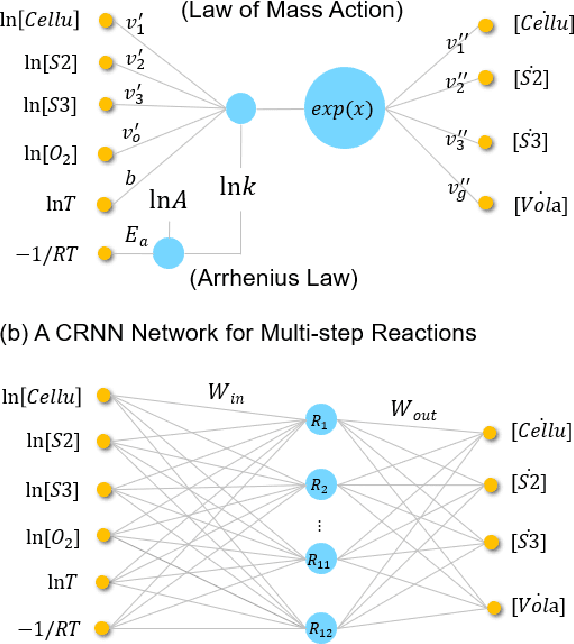
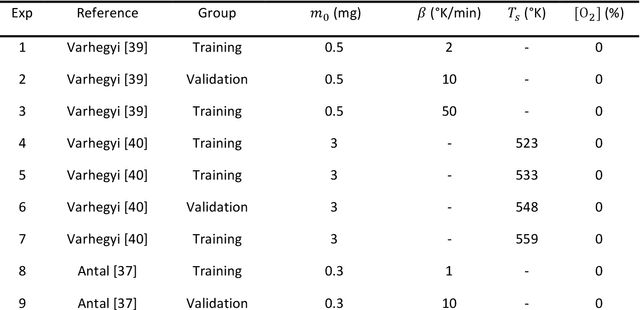
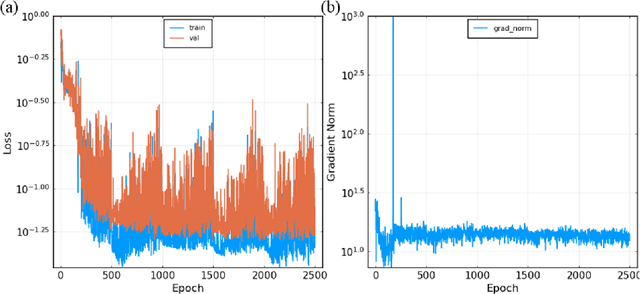
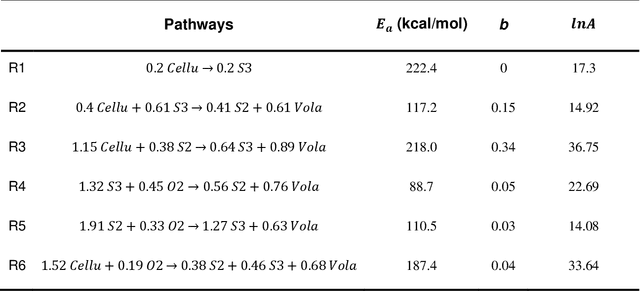
Modeling the burning processes of biomass such as wood, grass, and crops is crucial for the modeling and prediction of wildland and urban fire behavior. Despite its importance, the burning of solid fuels remains poorly understood, which can be partly attributed to the unknown chemical kinetics of most solid fuels. Most available kinetic models were built upon expert knowledge, which requires chemical insights and years of experience. This work presents a framework for autonomously discovering biomass pyrolysis kinetic models from thermogravimetric analyzer (TGA) experimental data using the recently developed chemical reaction neural networks (CRNN). The approach incorporated the CRNN model into the framework of neural ordinary differential equations to predict the residual mass in TGA data. In addition to the flexibility of neural-network-based models, the learned CRNN model is fully interpretable, by incorporating the fundamental physics laws, such as the law of mass action and Arrhenius law, into the neural network structure. The learned CRNN model can then be translated into the classical forms of biomass chemical kinetic models, which facilitates the extraction of chemical insights and the integration of the kinetic model into large-scale fire simulations. We demonstrated the effectiveness of the framework in predicting the pyrolysis and oxidation of cellulose. This successful demonstration opens the possibility of rapid and autonomous chemical kinetic modeling of solid fuels, such as wildfire fuels and industrial polymers.