 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStrategic priorities for transformative progress in advancing biology with proteomics and artificial intelligence
Feb 21, 2025

Artificial intelligence (AI) is transforming scientific research, including proteomics. Advances in mass spectrometry (MS)-based proteomics data quality, diversity, and scale, combined with groundbreaking AI techniques, are unlocking new challenges and opportunities in biological discovery. Here, we highlight key areas where AI is driving innovation, from data analysis to new biological insights. These include developing an AI-friendly ecosystem for proteomics data generation, sharing, and analysis; improving peptide and protein identification and quantification; characterizing protein-protein interactions and protein complexes; advancing spatial and perturbation proteomics; integrating multi-omics data; and ultimately enabling AI-empowered virtual cells.
Inference of cell dynamics on perturbation data using adjoint sensitivity
Apr 13, 2021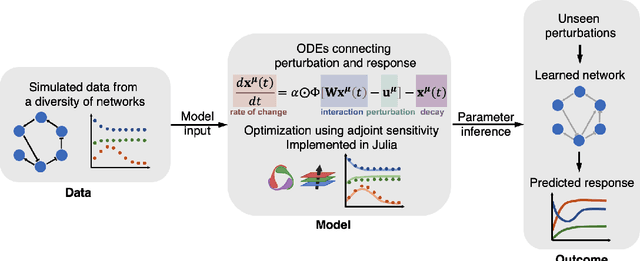
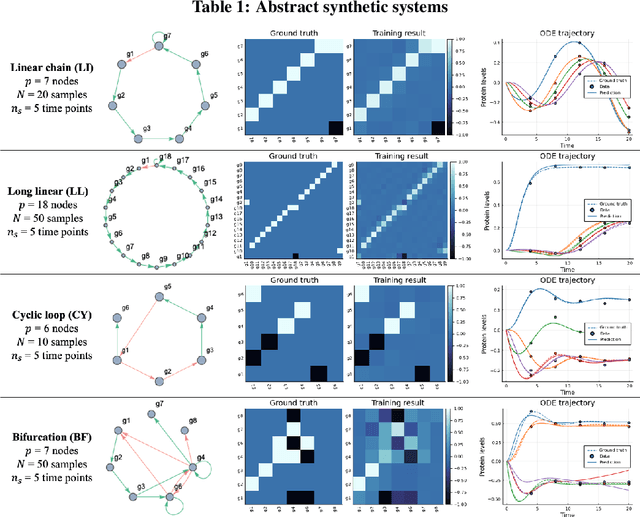
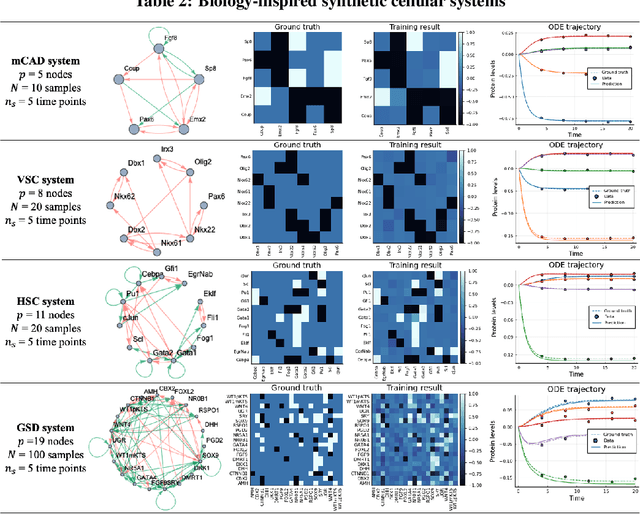
Data-driven dynamic models of cell biology can be used to predict cell response to unseen perturbations. Recent work (CellBox) had demonstrated the derivation of interpretable models with explicit interaction terms, in which the parameters were optimized using machine learning techniques. While the previous work was tested only in a single biological setting, this work aims to extend the range of applicability of this model inference approach to a diversity of biological systems. Here we adapted CellBox in Julia differential programming and augmented the method with adjoint algorithms, which has recently been used in the context of neural ODEs. We trained the models using simulated data from both abstract and biology-inspired networks, which afford the ability to evaluate the recovery of the ground truth network structure. The resulting accuracy of prediction by these models is high both in terms of low error against data and excellent agreement with the network structure used for the simulated training data. While there is no analogous ground truth for real life biological systems, this work demonstrates the ability to construct and parameterize a considerable diversity of network models with high predictive ability. The expectation is that this kind of procedure can be used on real perturbation-response data to derive models applicable to diverse biological systems.