 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA deep learning pipeline for cross-sectional and longitudinal multiview data integration
Dec 02, 2023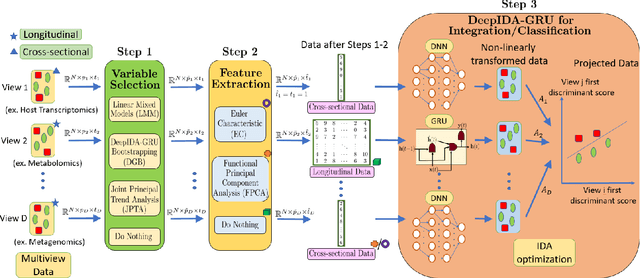



Biomedical research now commonly integrates diverse data types or views from the same individuals to better understand the pathobiology of complex diseases, but the challenge lies in meaningfully integrating these diverse views. Existing methods often require the same type of data from all views (cross-sectional data only or longitudinal data only) or do not consider any class outcome in the integration method, presenting limitations. To overcome these limitations, we have developed a pipeline that harnesses the power of statistical and deep learning methods to integrate cross-sectional and longitudinal data from multiple sources. Additionally, it identifies key variables contributing to the association between views and the separation among classes, providing deeper biological insights. This pipeline includes variable selection/ranking using linear and nonlinear methods, feature extraction using functional principal component analysis and Euler characteristics, and joint integration and classification using dense feed-forward networks and recurrent neural networks. We applied this pipeline to cross-sectional and longitudinal multi-omics data (metagenomics, transcriptomics, and metabolomics) from an inflammatory bowel disease (IBD) study and we identified microbial pathways, metabolites, and genes that discriminate by IBD status, providing information on the etiology of IBD. We conducted simulations to compare the two feature extraction methods. The proposed pipeline is available from the following GitHub repository: https://github.com/lasandrall/DeepIDA-GRU.
mvlearnR and Shiny App for multiview learning
Nov 25, 2023


The package mvlearnR and accompanying Shiny App is intended for integrating data from multiple sources or views or modalities (e.g. genomics, proteomics, clinical and demographic data). Most existing software packages for multiview learning are decentralized and offer limited capabilities, making it difficult for users to perform comprehensive integrative analysis. The new package wraps statistical and machine learning methods and graphical tools, providing a convenient and easy data integration workflow. For users with limited programming language, we provide a Shiny Application to facilitate data integration anywhere and on any device. The methods have potential to offer deeper insights into complex disease mechanisms. Availability and Implementation: mvlearnR is available from the following GitHub repository: https://github.com/lasandrall/mvlearnR. The web application is hosted on shinyapps.io and available at: https://multi-viewlearn.shinyapps.io/MultiView_Modeling/
Scalable Randomized Kernel Methods for Multiview Data Integration and Prediction
Apr 10, 2023



We develop scalable randomized kernel methods for jointly associating data from multiple sources and simultaneously predicting an outcome or classifying a unit into one of two or more classes. The proposed methods model nonlinear relationships in multiview data together with predicting a clinical outcome and are capable of identifying variables or groups of variables that best contribute to the relationships among the views. We use the idea that random Fourier bases can approximate shift-invariant kernel functions to construct nonlinear mappings of each view and we use these mappings and the outcome variable to learn view-independent low-dimensional representations. Through simulation studies, we show that the proposed methods outperform several other linear and nonlinear methods for multiview data integration. When the proposed methods were applied to gene expression, metabolomics, proteomics, and lipidomics data pertaining to COVID-19, we identified several molecular signatures forCOVID-19 status and severity. Results from our real data application and simulations with small sample sizes suggest that the proposed methods may be useful for small sample size problems. Availability: Our algorithms are implemented in Pytorch and interfaced in R and would be made available at: https://github.com/lasandrall/RandMVLearn.
Deep IDA: A Deep Learning Method for Integrative Discriminant Analysis of Multi-View Data with Feature Ranking -- An Application to COVID-19 severity
Nov 24, 2021
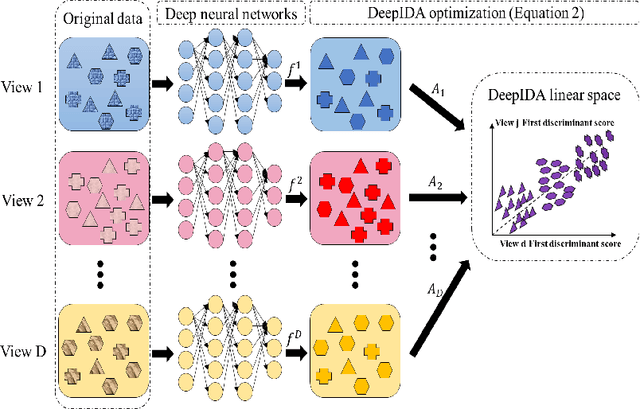

COVID-19 severity is due to complications from SARS-Cov-2 but the clinical course of the infection varies for individuals, emphasizing the need to better understand the disease at the molecular level. We use clinical and multiple molecular data (or views) obtained from patients with and without COVID-19 who were (or not) admitted to the intensive care unit to shed light on COVID-19 severity. Methods for jointly associating the views and separating the COVID-19 groups (i.e., one-step methods) have focused on linear relationships. The relationships between the views and COVID-19 patient groups, however, are too complex to be understood solely by linear methods. Existing nonlinear one-step methods cannot be used to identify signatures to aid in our understanding of the complexity of the disease. We propose Deep IDA (Integrative Discriminant Analysis) to address analytical challenges in our problem of interest. Deep IDA learns nonlinear projections of two or more views that maximally associate the views and separate the classes in each view, and permits feature ranking for interpretable findings. Our applications demonstrate that Deep IDA has competitive classification rates compared to other state-of-the-art methods and is able to identify molecular signatures that facilitate an understanding of COVID-19 severity.
sJIVE: Supervised Joint and Individual Variation Explained
Feb 26, 2021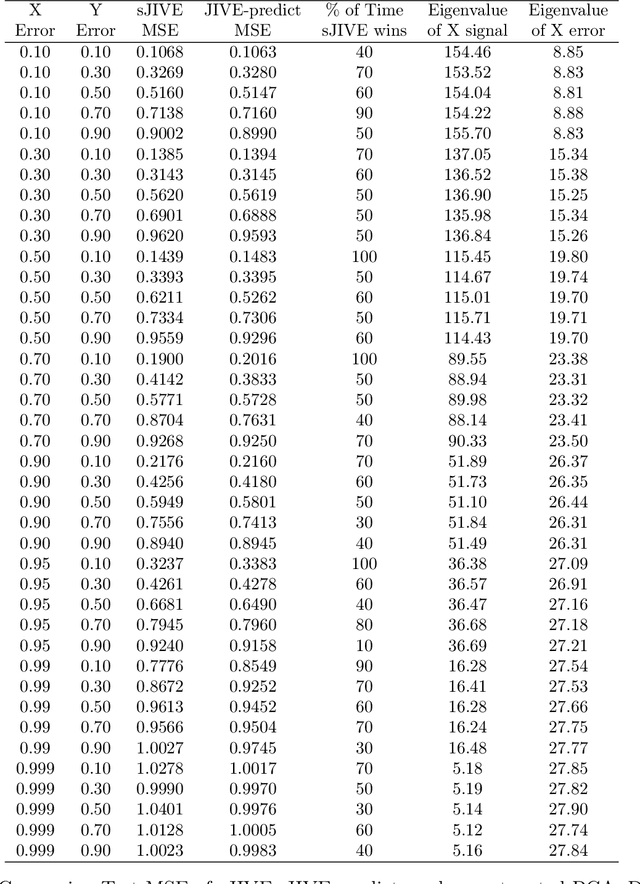


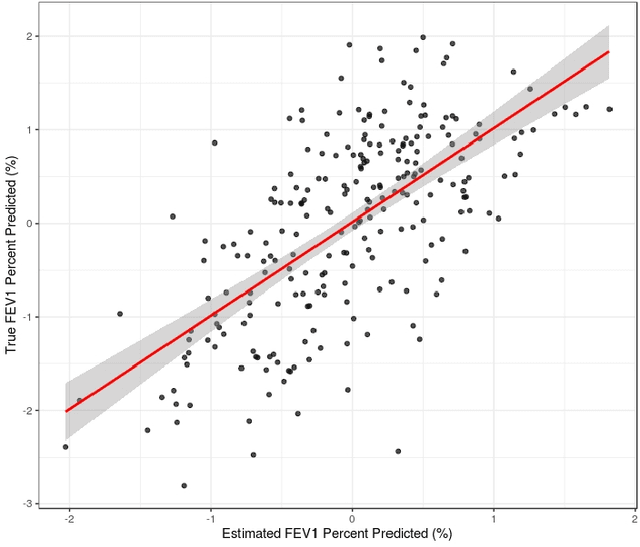
Analyzing multi-source data, which are multiple views of data on the same subjects, has become increasingly common in molecular biomedical research. Recent methods have sought to uncover underlying structure and relationships within and/or between the data sources, and other methods have sought to build a predictive model for an outcome using all sources. However, existing methods that do both are presently limited because they either (1) only consider data structure shared by all datasets while ignoring structures unique to each source, or (2) they extract underlying structures first without consideration to the outcome. We propose a method called supervised joint and individual variation explained (sJIVE) that can simultaneously (1) identify shared (joint) and source-specific (individual) underlying structure and (2) build a linear prediction model for an outcome using these structures. These two components are weighted to compromise between explaining variation in the multi-source data and in the outcome. Simulations show sJIVE to outperform existing methods when large amounts of noise are present in the multi-source data. An application to data from the COPDGene study reveals gene expression and proteomic patterns that are predictive of lung function. Functions to perform sJIVE are included in the R.JIVE package, available online at http://github.com/lockEF/r.jive .