 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultiple Augmented Reduced Rank Regression for Pan-Cancer Analysis
Aug 30, 2023



Statistical approaches that successfully combine multiple datasets are more powerful, efficient, and scientifically informative than separate analyses. To address variation architectures correctly and comprehensively for high-dimensional data across multiple sample sets (i.e., cohorts), we propose multiple augmented reduced rank regression (maRRR), a flexible matrix regression and factorization method to concurrently learn both covariate-driven and auxiliary structured variation. We consider a structured nuclear norm objective that is motivated by random matrix theory, in which the regression or factorization terms may be shared or specific to any number of cohorts. Our framework subsumes several existing methods, such as reduced rank regression and unsupervised multi-matrix factorization approaches, and includes a promising novel approach to regression and factorization of a single dataset (aRRR) as a special case. Simulations demonstrate substantial gains in power from combining multiple datasets, and from parsimoniously accounting for all structured variation. We apply maRRR to gene expression data from multiple cancer types (i.e., pan-cancer) from TCGA, with somatic mutations as covariates. The method performs well with respect to prediction and imputation of held-out data, and provides new insights into mutation-driven and auxiliary variation that is shared or specific to certain cancer types.
Deep IDA: A Deep Learning Method for Integrative Discriminant Analysis of Multi-View Data with Feature Ranking -- An Application to COVID-19 severity
Nov 24, 2021
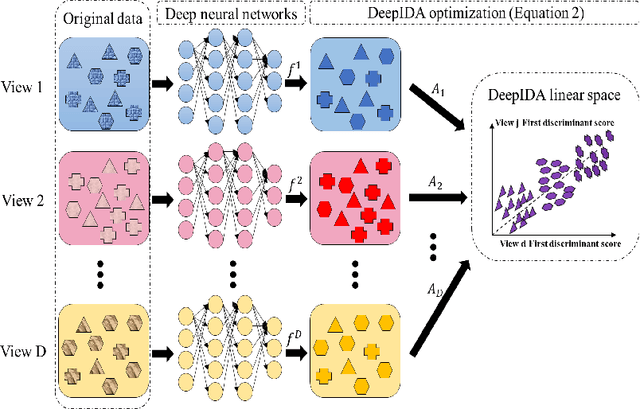

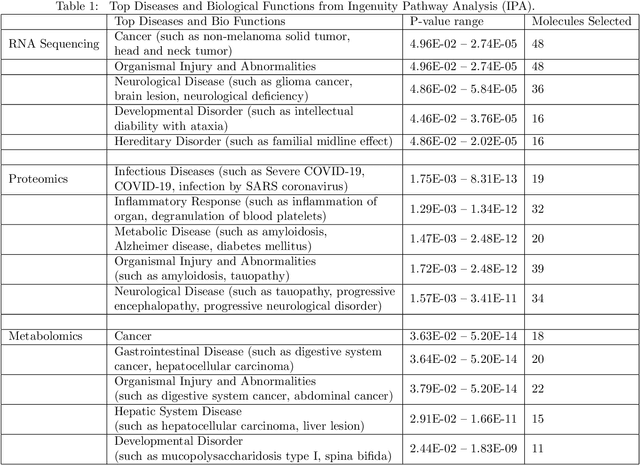
COVID-19 severity is due to complications from SARS-Cov-2 but the clinical course of the infection varies for individuals, emphasizing the need to better understand the disease at the molecular level. We use clinical and multiple molecular data (or views) obtained from patients with and without COVID-19 who were (or not) admitted to the intensive care unit to shed light on COVID-19 severity. Methods for jointly associating the views and separating the COVID-19 groups (i.e., one-step methods) have focused on linear relationships. The relationships between the views and COVID-19 patient groups, however, are too complex to be understood solely by linear methods. Existing nonlinear one-step methods cannot be used to identify signatures to aid in our understanding of the complexity of the disease. We propose Deep IDA (Integrative Discriminant Analysis) to address analytical challenges in our problem of interest. Deep IDA learns nonlinear projections of two or more views that maximally associate the views and separate the classes in each view, and permits feature ranking for interpretable findings. Our applications demonstrate that Deep IDA has competitive classification rates compared to other state-of-the-art methods and is able to identify molecular signatures that facilitate an understanding of COVID-19 severity.