 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWelQrate: Defining the Gold Standard in Small Molecule Drug Discovery Benchmarking
Nov 14, 2024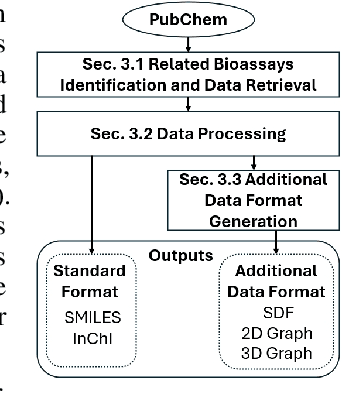
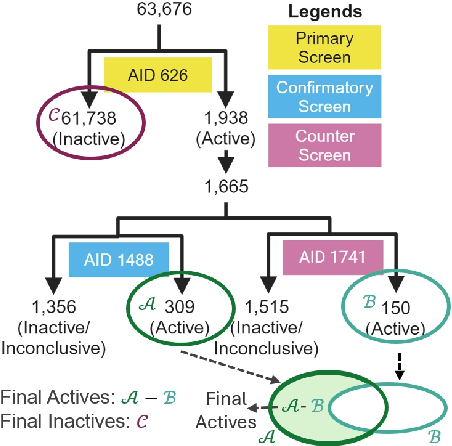
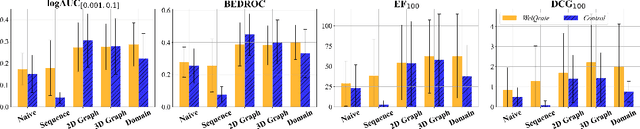
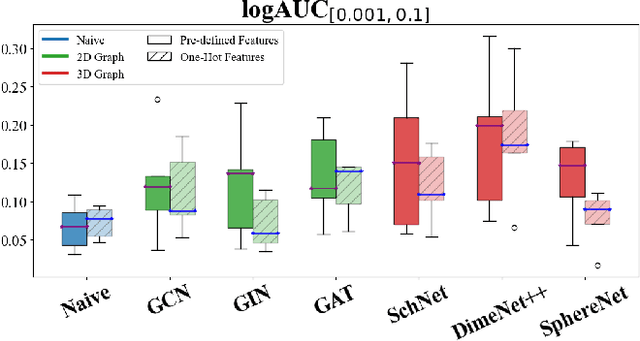
While deep learning has revolutionized computer-aided drug discovery, the AI community has predominantly focused on model innovation and placed less emphasis on establishing best benchmarking practices. We posit that without a sound model evaluation framework, the AI community's efforts cannot reach their full potential, thereby slowing the progress and transfer of innovation into real-world drug discovery. Thus, in this paper, we seek to establish a new gold standard for small molecule drug discovery benchmarking, WelQrate. Specifically, our contributions are threefold: WelQrate Dataset Collection - we introduce a meticulously curated collection of 9 datasets spanning 5 therapeutic target classes. Our hierarchical curation pipelines, designed by drug discovery experts, go beyond the primary high-throughput screen by leveraging additional confirmatory and counter screens along with rigorous domain-driven preprocessing, such as Pan-Assay Interference Compounds (PAINS) filtering, to ensure the high-quality data in the datasets; WelQrate Evaluation Framework - we propose a standardized model evaluation framework considering high-quality datasets, featurization, 3D conformation generation, evaluation metrics, and data splits, which provides a reliable benchmarking for drug discovery experts conducting real-world virtual screening; Benchmarking - we evaluate model performance through various research questions using the WelQrate dataset collection, exploring the effects of different models, dataset quality, featurization methods, and data splitting strategies on the results. In summary, we recommend adopting our proposed WelQrate as the gold standard in small molecule drug discovery benchmarking. The WelQrate dataset collection, along with the curation codes, and experimental scripts are all publicly available at WelQrate.org.
Deep Learning for Virtual Screening: Five Reasons to Use ROC Cost Functions
Jun 25, 2020Computer-aided drug discovery is an essential component of modern drug development. Therein, deep learning has become an important tool for rapid screening of billions of molecules in silico for potential hits containing desired chemical features. Despite its importance, substantial challenges persist in training these models, such as severe class imbalance, high decision thresholds, and lack of ground truth labels in some datasets. In this work we argue in favor of directly optimizing the receiver operating characteristic (ROC) in such cases, due to its robustness to class imbalance, its ability to compromise over different decision thresholds, certain freedom to influence the relative weights in this compromise, fidelity to typical benchmarking measures, and equivalence to positive/unlabeled learning. We also propose new training schemes (coherent mini-batch arrangement, and usage of out-of-batch samples) for cost functions based on the ROC, as well as a cost function based on the logAUC metric that facilitates early enrichment (i.e. improves performance at high decision thresholds, as often desired when synthesizing predicted hit compounds). We demonstrate that these approaches outperform standard deep learning approaches on a series of PubChem high-throughput screening datasets that represent realistic and diverse drug discovery campaigns on major drug target families.