 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEquiBoost: An Equivariant Boosting Approach to Molecular Conformation Generation
Jan 09, 2025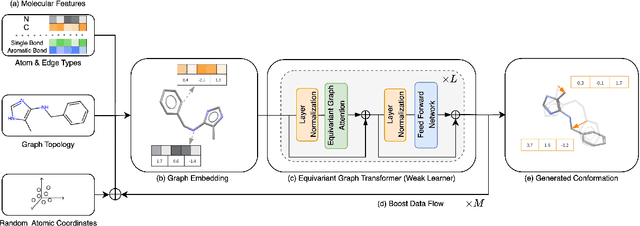
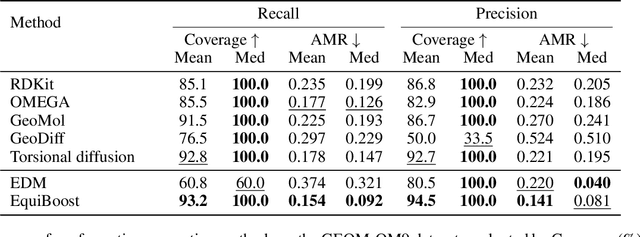
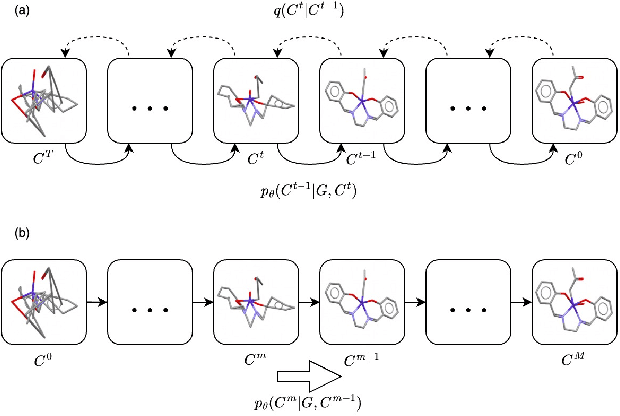
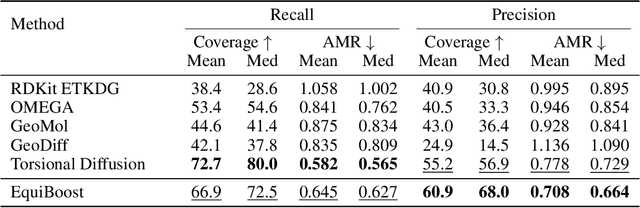
Molecular conformation generation plays key roles in computational drug design. Recently developed deep learning methods, particularly diffusion models have reached competitive performance over traditional cheminformatical approaches. However, these methods are often time-consuming or require extra support from traditional methods. We propose EquiBoost, a boosting model that stacks several equivariant graph transformers as weak learners, to iteratively refine 3D conformations of molecules. Without relying on diffusion techniques, EquiBoost balances accuracy and efficiency more effectively than diffusion-based methods. Notably, compared to the previous state-of-the-art diffusion method, EquiBoost improves generation quality and preserves diversity, achieving considerably better precision of Average Minimum RMSD (AMR) on the GEOM datasets. This work rejuvenates boosting and sheds light on its potential to be a robust alternative to diffusion models in certain scenarios.
EquiFlow: Equivariant Conditional Flow Matching with Optimal Transport for 3D Molecular Conformation Prediction
Dec 15, 2024
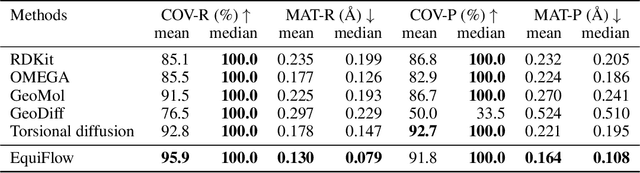


Molecular 3D conformations play a key role in determining how molecules interact with other molecules or protein surfaces. Recent deep learning advancements have improved conformation prediction, but slow training speeds and difficulties in utilizing high-degree features limit performance. We propose EquiFlow, an equivariant conditional flow matching model with optimal transport. EquiFlow uniquely applies conditional flow matching in molecular 3D conformation prediction, leveraging simulation-free training to address slow training speeds. It uses a modified Equiformer model to encode Cartesian molecular conformations along with their atomic and bond properties into higher-degree embeddings. Additionally, EquiFlow employs an ODE solver, providing faster inference speeds compared to diffusion models with SDEs. Experiments on the QM9 dataset show that EquiFlow predicts small molecule conformations more accurately than current state-of-the-art models.
M4: Multi-Proxy Multi-Gate Mixture of Experts Network for Multiple Instance Learning in Histopathology Image Analysis
Jul 24, 2024



Multiple instance learning (MIL) has been successfully applied for whole slide images (WSIs) analysis in computational pathology, enabling a wide range of prediction tasks from tumor subtyping to inferring genetic mutations and multi-omics biomarkers. However, existing MIL methods predominantly focus on single-task learning, resulting in not only overall low efficiency but also the overlook of inter-task relatedness. To address these issues, we proposed an adapted architecture of Multi-gate Mixture-of-experts with Multi-proxy for Multiple instance learning (M4), and applied this framework for simultaneous prediction of multiple genetic mutations from WSIs. The proposed M4 model has two main innovations: (1) utilizing a mixture of experts with multiple gating strategies for multi-genetic mutation prediction on a single pathological slide; (2) constructing multi-proxy expert network and gate network for comprehensive and effective modeling of pathological image information. Our model achieved significant improvements across five tested TCGA datasets in comparison to current state-of-the-art single-task methods. The code is available at:https://github.com/Bigyehahaha/M4.