 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePointIso: Point Cloud Based Deep Learning Model for Detecting Arbitrary-Precision Peptide Features in LC-MS Map through Attention Based Segmentation
Sep 15, 2020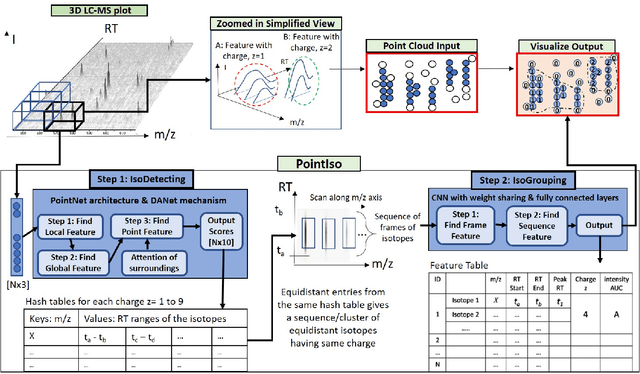


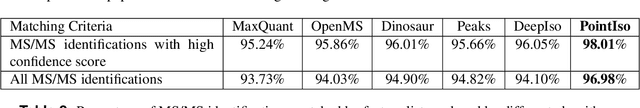
A promising technique of discovering disease biomarkers is to measure the relative protein abundance in multiple biofluid samples through liquid chromatography with tandem mass spectrometry (LC-MS/MS) based quantitative proteomics. The key step involves peptide feature detection in LC-MS map, along with its charge and intensity. Existing heuristic algorithms suffer from inaccurate parameters since different settings of the parameters result in significantly different outcomes. Therefore, we propose PointIso, to serve the necessity of an automated system for peptide feature detection that is able to find out the proper parameters itself, and is easily adaptable to different types of datasets. It consists of an attention based scanning step for segmenting the multi-isotopic pattern of peptide features along with charge and a sequence classification step for grouping those isotopes into potential peptide features. PointIso is the first point cloud based, arbitrary-precision deep learning network to address the problem and achieves 98% detection of high quality MS/MS identifications in a benchmark dataset, which is higher than several other widely used algorithms. Besides contributing to the proteomics study, we believe our novel segmentation technique should serve the general image processing domain as well.
DeepNovoV2: Better de novo peptide sequencing with deep learning
May 22, 2019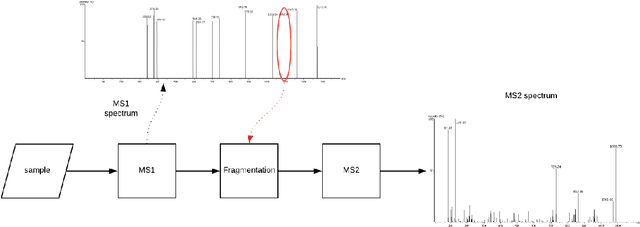
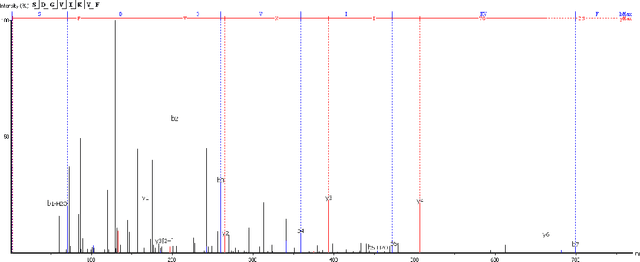
Personalized cancer vaccines are envisioned as the next generation rational cancer immunotherapy. The key step in developing personalized therapeutic cancer vaccines is to identify tumor-specific neoantigens that are on the surface of tumor cells. A promising method for this is through de novo peptide sequencing from mass spectrometry data. In this paper we introduce DeepNovoV2, the state-of-the-art model for peptide sequencing. In DeepNovoV2, a spectrum is directly represented as a set of (m/z, intensity) pairs, therefore it does not suffer from the accuracy-speed/memory trade-off problem. The model combines an order invariant network structure (T-Net) and recurrent neural networks and provides a complete end-to-end training and prediction framework to sequence patterns of peptides. Our experiments on a wide variety of data from different species show that DeepNovoV2 outperforms previous state-of-the-art methods, achieving 13.01-23.95\% higher accuracy at the peptide level.
DeepIso: A Deep Learning Model for Peptide Feature Detection
Dec 09, 2017

Liquid chromatography with tandem mass spectrometry (LC-MS/MS) based proteomics is a well-established research field with major applications such as identification of disease biomarkers, drug discovery, drug design and development. In proteomics, protein identification and quantification is a fundamental task, which is done by first enzymatically digesting it into peptides, and then analyzing peptides by LC-MS/MS instruments. The peptide feature detection and quantification from an LC-MS map is the first step in typical analysis workflows. In this paper we propose a novel deep learning based model, DeepIso, that uses Convolutional Neural Networks (CNNs) to scan an LC-MS map to detect peptide features and estimate their abundance. Existing tools are often designed with limited engineered features based on domain knowledge, and depend on pretrained parameters which are hardly updated despite huge amount of new coming proteomic data. Our proposed model, on the other hand, is capable of learning multiple levels of representation of high dimensional data through its many layers of neurons and continuously evolving with newly acquired data. To evaluate our proposed model, we use an antibody dataset including a heavy and a light chain, each digested by Asp-N, Chymotrypsin, Trypsin, thus giving six LC-MS maps for the experiment. Our model achieves 93.21% sensitivity with specificity of 99.44% on this dataset. Our results demonstrate that novel deep learning tools are desirable to advance the state-of-the-art in protein identification and quantification.
Protein identification with deep learning: from abc to xyz
Oct 08, 2017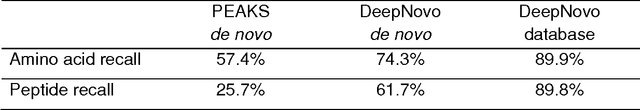
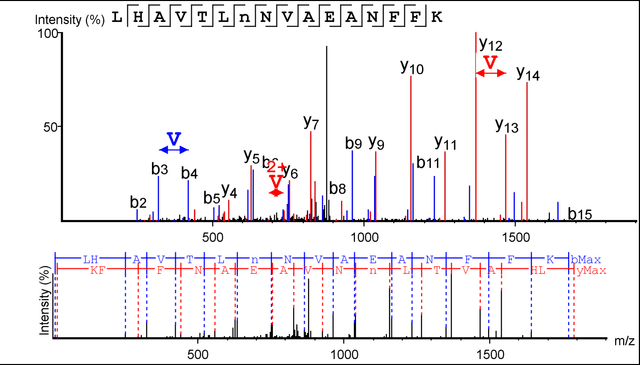

Proteins are the main workhorses of biological functions in a cell, a tissue, or an organism. Identification and quantification of proteins in a given sample, e.g. a cell type under normal/disease conditions, are fundamental tasks for the understanding of human health and disease. In this paper, we present DeepNovo, a deep learning-based tool to address the problem of protein identification from tandem mass spectrometry data. The idea was first proposed in the context of de novo peptide sequencing [1] in which convolutional neural networks and recurrent neural networks were applied to predict the amino acid sequence of a peptide from its spectrum, a similar task to generating a caption from an image. We further develop DeepNovo to perform sequence database search, the main technique for peptide identification that greatly benefits from numerous existing protein databases. We combine two modules de novo sequencing and database search into a single deep learning framework for peptide identification, and integrate de Bruijn graph assembly technique to offer a complete solution to reconstruct protein sequences from tandem mass spectrometry data. This paper describes a comprehensive protocol of DeepNovo for protein identification, including training neural network models, dynamic programming search, database querying, estimation of false discovery rate, and de Bruijn graph assembly. Training and testing data, model implementations, and comprehensive tutorials in form of IPython notebooks are available in our GitHub repository (https://github.com/nh2tran/DeepNovo).