 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExplainable Molecular Property Prediction: Aligning Chemical Concepts with Predictions via Language Models
May 25, 2024



Providing explainable molecule property predictions is critical for many scientific domains, such as drug discovery and material science. Though transformer-based language models have shown great potential in accurate molecular property prediction, they neither provide chemically meaningful explanations nor faithfully reveal the molecular structure-property relationships. In this work, we develop a new framework for explainable molecular property prediction based on language models, dubbed as Lamole, which can provide chemical concepts-aligned explanations. We first leverage a designated molecular representation -- the Group SELFIES -- as it can provide chemically meaningful semantics. Because attention mechanisms in Transformers can inherently capture relationships within the input, we further incorporate the attention weights and gradients together to generate explanations for capturing the functional group interactions. We then carefully craft a marginal loss to explicitly optimize the explanations to be able to align with the chemists' annotations. We bridge the manifold hypothesis with the elaborated marginal loss to prove that the loss can align the explanations with the tangent space of the data manifold, leading to concept-aligned explanations. Experimental results over six mutagenicity datasets and one hepatotoxicity dataset demonstrate Lamole can achieve comparable classification accuracy and boost the explanation accuracy by up to 14.8%, being the state-of-the-art in explainable molecular property prediction.
Graph Convolutional Neural Networks for Polymers Property Prediction
Nov 15, 2018
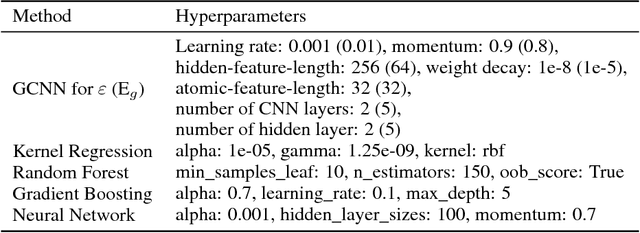


A fast and accurate predictive tool for polymer properties is demanding and will pave the way to iterative inverse design. In this work, we apply graph convolutional neural networks (GCNN) to predict the dielectric constant and energy bandgap of polymers. Using density functional theory (DFT) calculated properties as the ground truth, GCNN can achieve remarkable agreement with DFT results. Moreover, we show that GCNN outperforms other machine learning algorithms. Our work proves that GCNN relies only on morphological data of polymers and removes the requirement for complicated hand-crafted descriptors, while still offering accuracy in fast predictions.