 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVerSe: A Vertebrae Labelling and Segmentation Benchmark
Jan 24, 2020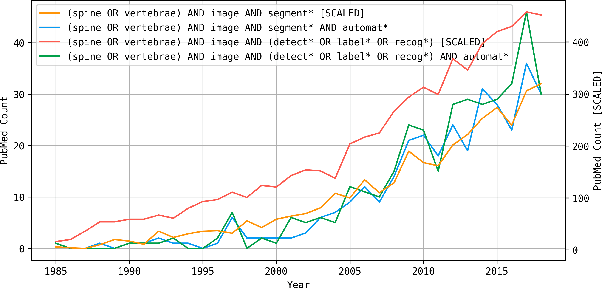
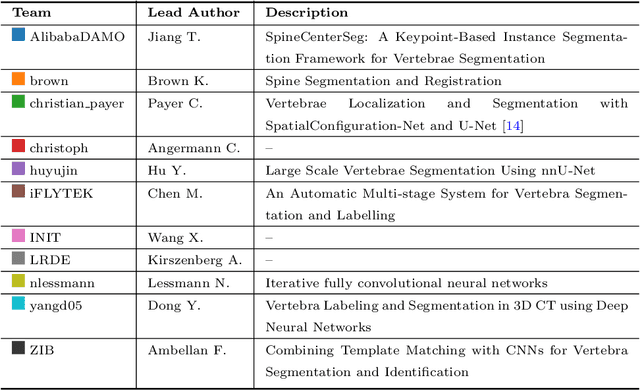
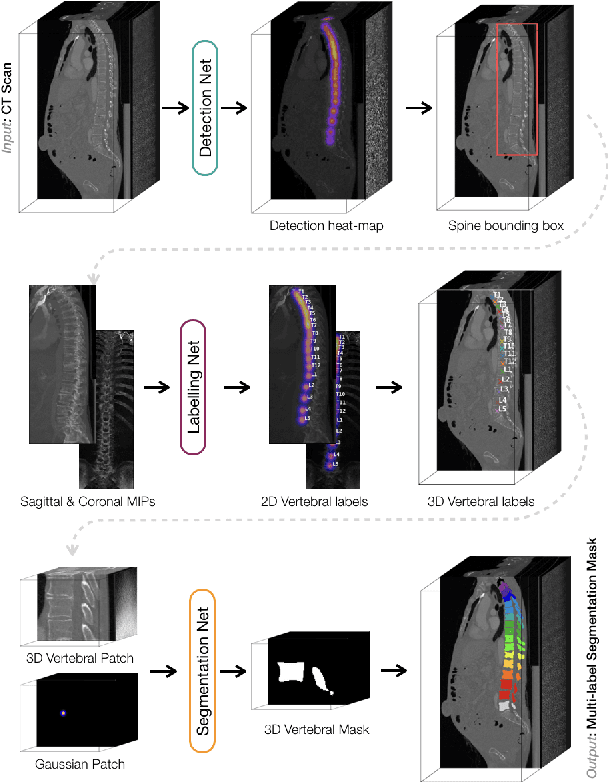
In this paper we report the challenge set-up and results of the Large Scale Vertebrae Segmentation Challenge (VerSe) organized in conjunction with the MICCAI 2019. The challenge consisted of two tasks, vertebrae labelling and vertebrae segmentation. For this a total of 160 multidetector CT scan cohort closely resembling clinical setting was prepared and was annotated at a voxel-level by a human-machine hybrid algorithm. In this paper we also present the annotation protocol and the algorithm that aided the medical experts in the annotation process. Eleven fully automated algorithms were benchmarked on this data with the best performing algorithm achieving a vertebrae identification rate of 95% and a Dice coefficient of 90%. VerSe'19 is an open-call challenge at its image data along with the annotations and evaluation tools will continue to be publicly accessible through its online portal.
Evolutionary distances in the twilight zone -- a rational kernel approach
Nov 23, 2010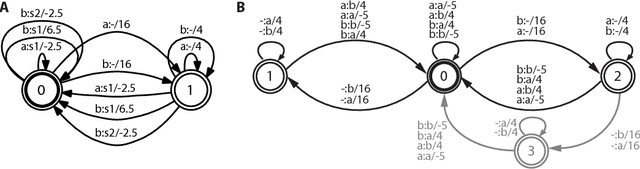
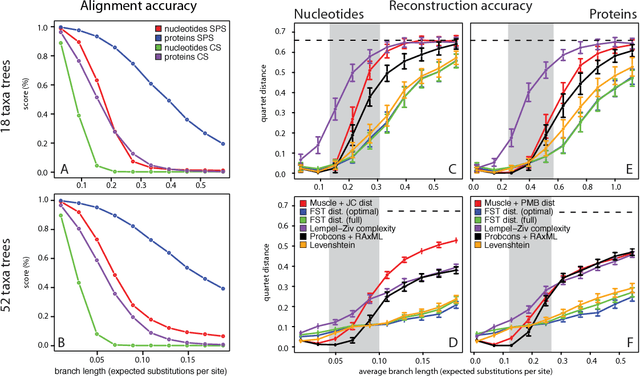
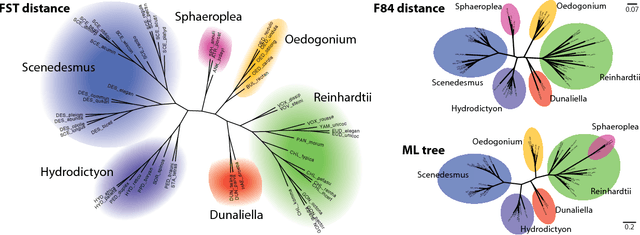
Phylogenetic tree reconstruction is traditionally based on multiple sequence alignments (MSAs) and heavily depends on the validity of this information bottleneck. With increasing sequence divergence, the quality of MSAs decays quickly. Alignment-free methods, on the other hand, are based on abstract string comparisons and avoid potential alignment problems. However, in general they are not biologically motivated and ignore our knowledge about the evolution of sequences. Thus, it is still a major open question how to define an evolutionary distance metric between divergent sequences that makes use of indel information and known substitution models without the need for a multiple alignment. Here we propose a new evolutionary distance metric to close this gap. It uses finite-state transducers to create a biologically motivated similarity score which models substitutions and indels, and does not depend on a multiple sequence alignment. The sequence similarity score is defined in analogy to pairwise alignments and additionally has the positive semi-definite property. We describe its derivation and show in simulation studies and real-world examples that it is more accurate in reconstructing phylogenies than competing methods. The result is a new and accurate way of determining evolutionary distances in and beyond the twilight zone of sequence alignments that is suitable for large datasets.
* to appear in PLoS ONE