 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRobots Learning to Say `No': Prohibition and Rejective Mechanisms in Acquisition of Linguistic Negation
Oct 28, 2018
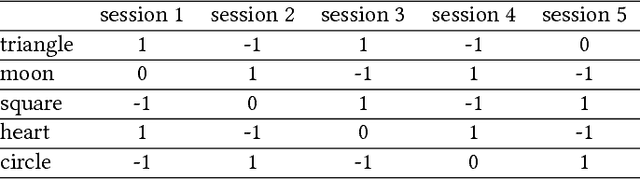
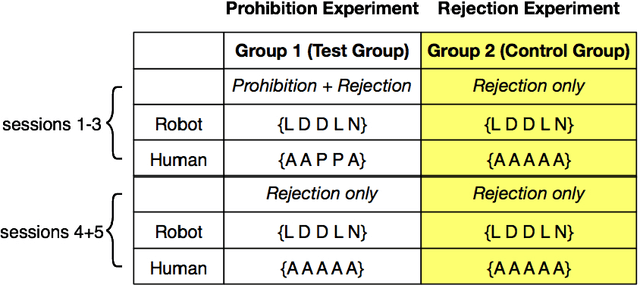
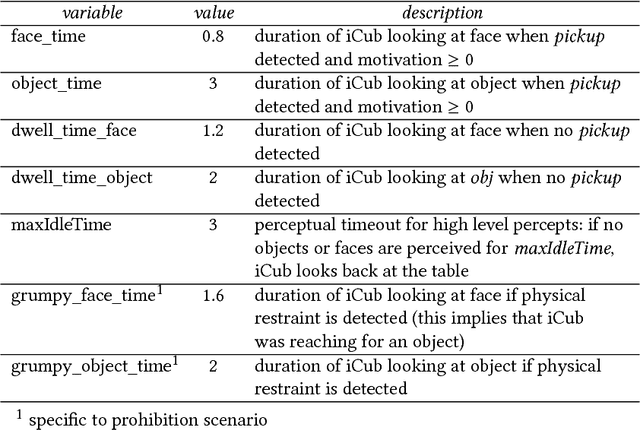
`No' belongs to the first ten words used by children and embodies the first active form of linguistic negation. Despite its early occurrence the details of its acquisition process remain largely unknown. The circumstance that `no' cannot be construed as a label for perceptible objects or events puts it outside of the scope of most modern accounts of language acquisition. Moreover, most symbol grounding architectures will struggle to ground the word due to its non-referential character. In an experimental study involving the child-like humanoid robot iCub that was designed to illuminate the acquisition process of negation words, the robot is deployed in several rounds of speech-wise unconstrained interaction with na\"ive participants acting as its language teachers. The results corroborate the hypothesis that affect or volition plays a pivotal role in the socially distributed acquisition process. Negation words are prosodically salient within prohibitive utterances and negative intent interpretations such that they can be easily isolated from the teacher's speech signal. These words subsequently may be grounded in negative affective states. However, observations of the nature of prohibitive acts and the temporal relationships between its linguistic and extra-linguistic components raise serious questions over the suitability of Hebbian-type algorithms for language grounding.
Evolutionary distances in the twilight zone -- a rational kernel approach
Nov 23, 2010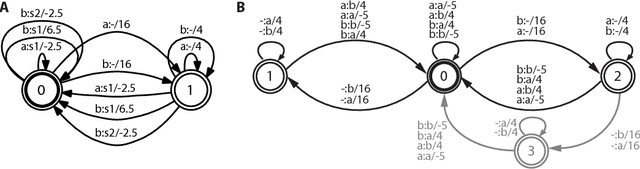
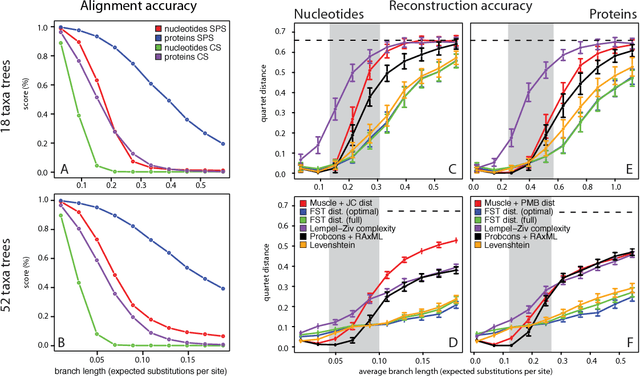
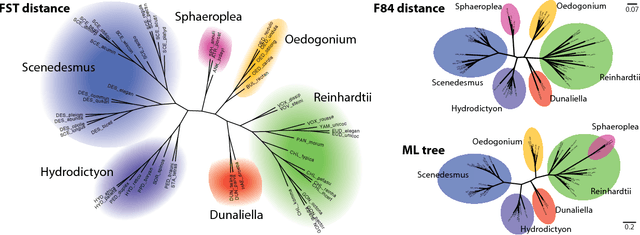
Phylogenetic tree reconstruction is traditionally based on multiple sequence alignments (MSAs) and heavily depends on the validity of this information bottleneck. With increasing sequence divergence, the quality of MSAs decays quickly. Alignment-free methods, on the other hand, are based on abstract string comparisons and avoid potential alignment problems. However, in general they are not biologically motivated and ignore our knowledge about the evolution of sequences. Thus, it is still a major open question how to define an evolutionary distance metric between divergent sequences that makes use of indel information and known substitution models without the need for a multiple alignment. Here we propose a new evolutionary distance metric to close this gap. It uses finite-state transducers to create a biologically motivated similarity score which models substitutions and indels, and does not depend on a multiple sequence alignment. The sequence similarity score is defined in analogy to pairwise alignments and additionally has the positive semi-definite property. We describe its derivation and show in simulation studies and real-world examples that it is more accurate in reconstructing phylogenies than competing methods. The result is a new and accurate way of determining evolutionary distances in and beyond the twilight zone of sequence alignments that is suitable for large datasets.
* to appear in PLoS ONE