 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMamba State-Space Models Can Be Strong Downstream Learners
May 31, 2024Mamba state-space models (SSMs) have recently outperformed state-of-the-art (SOTA) Transformer large language models (LLMs) in various tasks and been widely adapted. However, Mamba's downstream learning capabilities remain either unexplored$\unicode{x2013}$e.g., mixed-precision (MPFT) and parameter-efficient fine-tuning (PEFT)--or under-evaluated$\unicode{x2013}$e.g., in-context learning (ICL). For the latter, recent works reported Mamba's ICL rivals SOTA Transformer LLMs using non-standard benchmarks. In contrast, we show that on standard benchmarks, pretrained Mamba models achieve only 38% of the ICL performance improvements (over zero-shot) of comparable Transformers. Enabling MPFT and PEFT in Mamba architectures is challenging due to recurrent dynamics and highly customized CUDA kernels, respectively. However, we prove that Mamba's recurrent dynamics are robust to small input changes using dynamical systems theory. Empirically, we show that performance changes in Mamba's inference and fine-tuning due to mixed-precision align with Transformer LLMs. Furthermore, we show that targeting key memory buffers in Mamba's customized CUDA kernels for low-rank adaptation regularizes SSM parameters, thus achieving parameter efficiency while retaining speedups. We show that combining MPFT and PEFT enables up to 2.15 times more tokens-per-second and 65.5% reduced per-token-memory compared to full Mamba fine-tuning, while achieving up to 81.5% of the ICL performance improvements (over zero-shot) of comparably fine-tuned Transformers.
GPU-Accelerated Primal Learning for Extremely Fast Large-Scale Classification
Aug 08, 2020

One of the most efficient methods to solve L2-regularized primal problems, such as logistic regression and linear support vector machine (SVM) classification, is the widely used trust region Newton algorithm, TRON. While TRON has recently been shown to enjoy substantial speedups on shared-memory multi-core systems, exploiting graphical processing units (GPUs) to speed up the method is significantly more difficult, owing to the highly complex and heavily sequential nature of the algorithm. In this work, we show that using judicious GPU-optimization principles, TRON training time for different losses and feature representations may be drastically reduced. For sparse feature sets, we show that using GPUs to train logistic regression classifiers in LIBLINEAR is up to an order-of-magnitude faster than solely using multithreading. For dense feature sets--which impose far more stringent memory constraints--we show that GPUs substantially reduce the lengthy SVM learning times required for state-of-the-art proteomics analysis, leading to dramatic improvements over recently proposed speedups. Furthermore, we show how GPU speedups may be mixed with multithreading to enable such speedups when the dataset is too large for GPU memory requirements; on a massive dense proteomics dataset of nearly a quarter-billion data instances, these mixed-architecture speedups reduce SVM analysis time from over half a week to less than a single day while using limited GPU memory.
Learning Concave Conditional Likelihood Models for Improved Analysis of Tandem Mass Spectra
Sep 04, 2019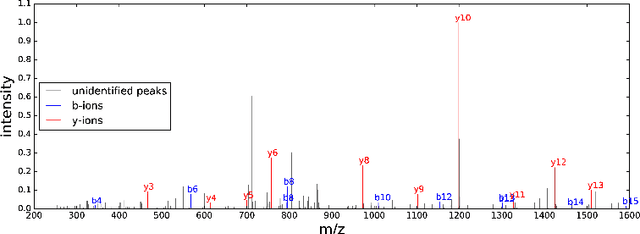
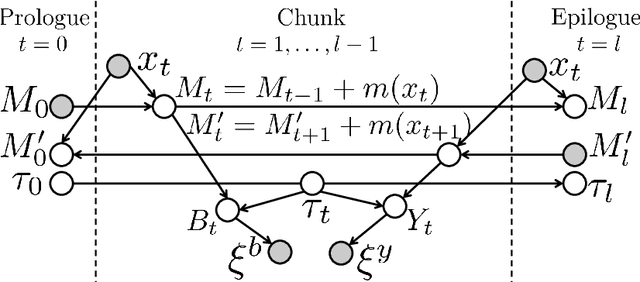
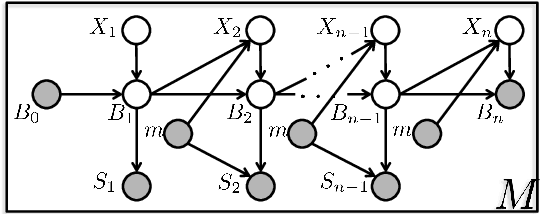
The most widely used technology to identify the proteins present in a complex biological sample is tandem mass spectrometry, which quickly produces a large collection of spectra representative of the peptides (i.e., protein subsequences) present in the original sample. In this work, we greatly expand the parameter learning capabilities of a dynamic Bayesian network (DBN) peptide-scoring algorithm, Didea, by deriving emission distributions for which its conditional log-likelihood scoring function remains concave. We show that this class of emission distributions, called Convex Virtual Emissions (CVEs), naturally generalizes the log-sum-exp function while rendering both maximum likelihood estimation and conditional maximum likelihood estimation concave for a wide range of Bayesian networks. Utilizing CVEs in Didea allows efficient learning of a large number of parameters while ensuring global convergence, in stark contrast to Didea's previous parameter learning framework (which could only learn a single parameter using a costly grid search) and other trainable models (which only ensure convergence to local optima). The newly trained scoring function substantially outperforms the state-of-the-art in both scoring function accuracy and downstream Fisher kernel analysis. Furthermore, we significantly improve Didea's runtime performance through successive optimizations to its message passing schedule and derive explicit connections between Didea's new concave score and related MS/MS scoring functions.
Gradients of Generative Models for Improved Discriminative Analysis of Tandem Mass Spectra
Sep 04, 2019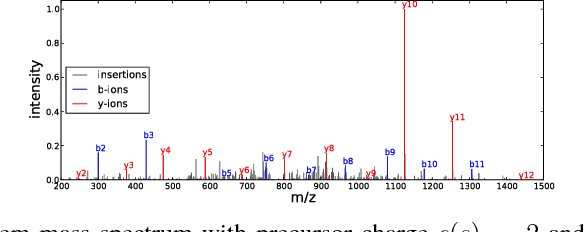
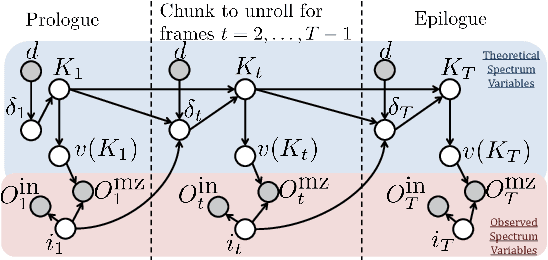
Tandem mass spectrometry (MS/MS) is a high-throughput technology used toidentify the proteins in a complex biological sample, such as a drop of blood. A collection of spectra is generated at the output of the process, each spectrum of which is representative of a peptide (protein subsequence) present in the original complex sample. In this work, we leverage the log-likelihood gradients of generative models to improve the identification of such spectra. In particular, we show that the gradient of a recently proposed dynamic Bayesian network (DBN) may be naturally employed by a kernel-based discriminative classifier. The resulting Fisher kernel substantially improves upon recent attempts to combine generative and discriminative models for post-processing analysis, outperforming all other methods on the evaluated datasets. We extend the improved accuracy offered by the Fisher kernel framework to other search algorithms by introducing Theseus, a DBN representing a large number of widely used MS/MS scoring functions. Furthermore, with gradient ascent and max-product inference at hand, we use Theseus to learn model parameters without any supervision.
Jensen: An Easily-Extensible C++ Toolkit for Production-Level Machine Learning and Convex Optimization
Jul 17, 2018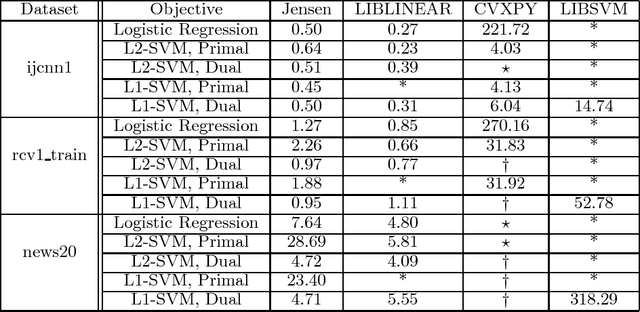
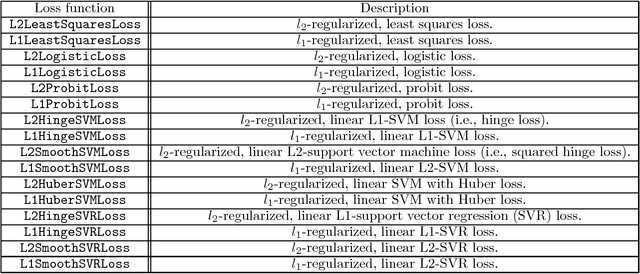
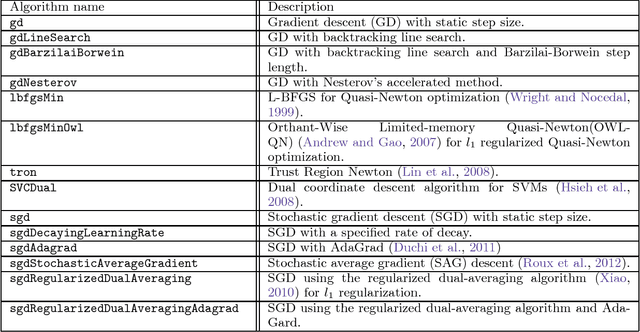
This paper introduces Jensen, an easily extensible and scalable toolkit for production-level machine learning and convex optimization. Jensen implements a framework of convex (or loss) functions, convex optimization algorithms (including Gradient Descent, L-BFGS, Stochastic Gradient Descent, Conjugate Gradient, etc.), and a family of machine learning classifiers and regressors (Logistic Regression, SVMs, Least Square Regression, etc.). This framework makes it possible to deploy and train models with a few lines of code, and also extend and build upon this by integrating new loss functions and optimization algorithms.
Faster graphical model identification of tandem mass spectra using peptide word lattices
Oct 29, 2014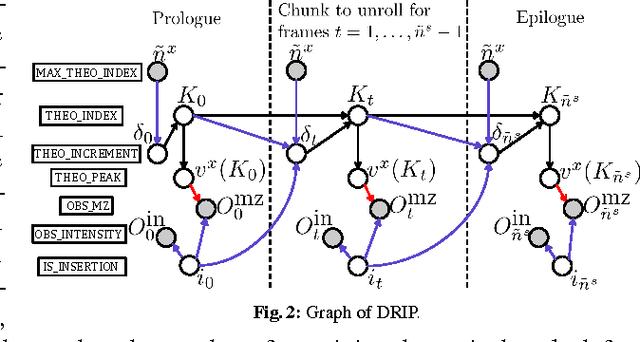

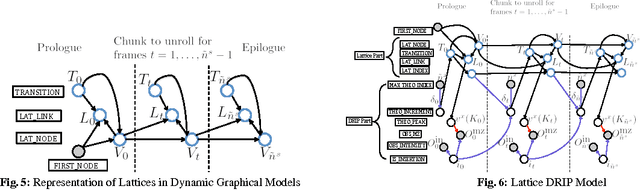
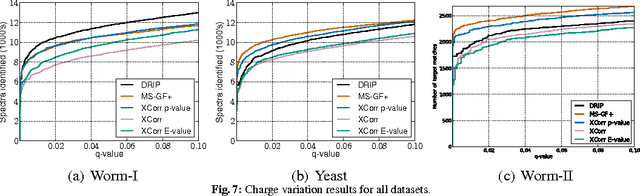
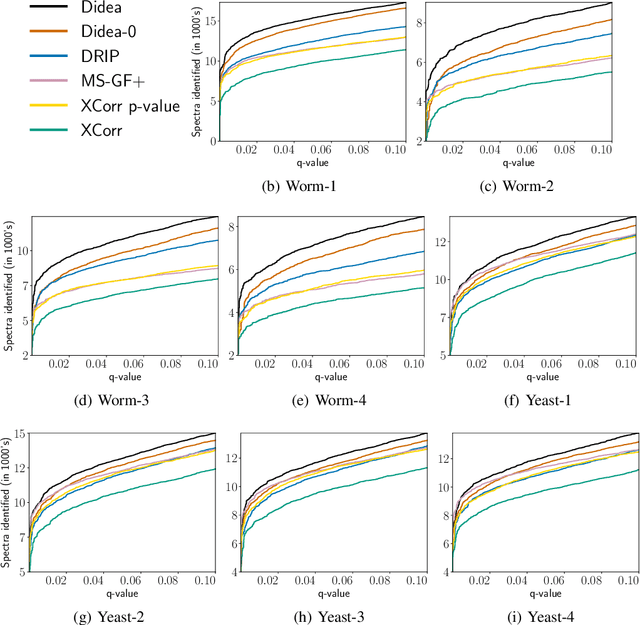
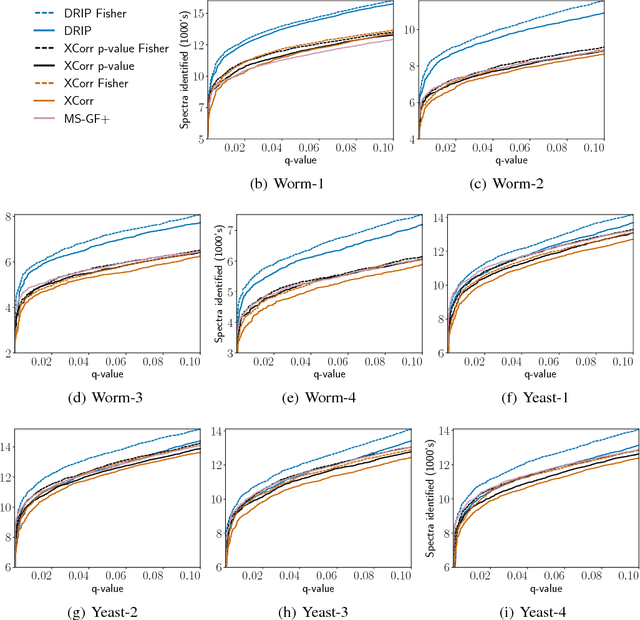
Liquid chromatography coupled with tandem mass spectrometry, also known as shotgun proteomics, is a widely-used high-throughput technology for identifying proteins in complex biological samples. Analysis of the tens of thousands of fragmentation spectra produced by a typical shotgun proteomics experiment begins by assigning to each observed spectrum the peptide hypothesized to be responsible for generating the spectrum, typically done by searching each spectrum against a database of peptides. We have recently described a machine learning method---Dynamic Bayesian Network for Rapid Identification of Peptides (DRIP)---that not only achieves state-of-the-art spectrum identification performance on a variety of datasets but also provides a trainable model capable of returning valuable auxiliary information regarding specific peptide-spectrum matches. In this work, we present two significant improvements to DRIP. First, we describe how to use word lattices, which are widely used in natural language processing, to significantly speed up DRIP's computations. To our knowledge, all existing shotgun proteomics search engines compute independent scores between a given observed spectrum and each possible candidate peptide from the database. The key idea of the word lattice is to represent the set of candidate peptides in a single data structure, thereby allowing sharing of redundant computations among the different candidates. We demonstrate that using lattices in conjunction with DRIP leads to speedups on the order of tens across yeast and worm data sets. Second, we introduce a variant of DRIP that uses a discriminative training framework, performing maximum mutual entropy estimation rather than maximum likelihood estimation. This modification improves DRIP's statistical power, enabling us to increase the number of identified spectrum at a 1% false discovery rate on yeast and worm data sets.