 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFaster graphical model identification of tandem mass spectra using peptide word lattices
Oct 29, 2014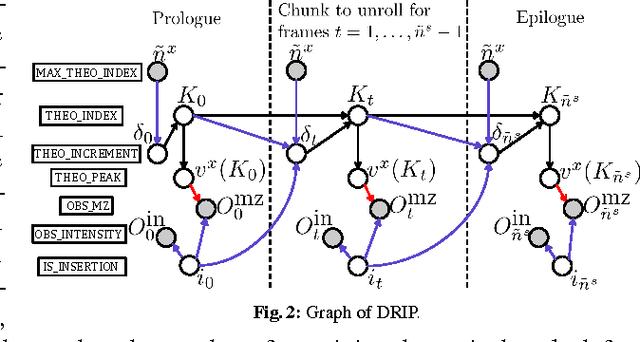

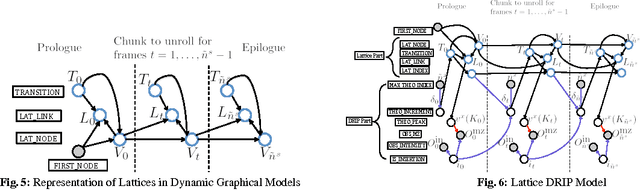
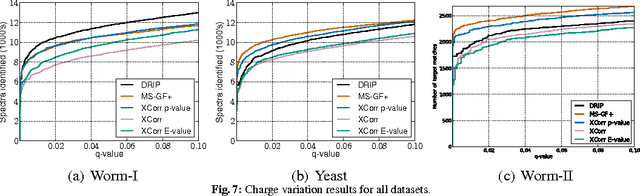
Liquid chromatography coupled with tandem mass spectrometry, also known as shotgun proteomics, is a widely-used high-throughput technology for identifying proteins in complex biological samples. Analysis of the tens of thousands of fragmentation spectra produced by a typical shotgun proteomics experiment begins by assigning to each observed spectrum the peptide hypothesized to be responsible for generating the spectrum, typically done by searching each spectrum against a database of peptides. We have recently described a machine learning method---Dynamic Bayesian Network for Rapid Identification of Peptides (DRIP)---that not only achieves state-of-the-art spectrum identification performance on a variety of datasets but also provides a trainable model capable of returning valuable auxiliary information regarding specific peptide-spectrum matches. In this work, we present two significant improvements to DRIP. First, we describe how to use word lattices, which are widely used in natural language processing, to significantly speed up DRIP's computations. To our knowledge, all existing shotgun proteomics search engines compute independent scores between a given observed spectrum and each possible candidate peptide from the database. The key idea of the word lattice is to represent the set of candidate peptides in a single data structure, thereby allowing sharing of redundant computations among the different candidates. We demonstrate that using lattices in conjunction with DRIP leads to speedups on the order of tens across yeast and worm data sets. Second, we introduce a variant of DRIP that uses a discriminative training framework, performing maximum mutual entropy estimation rather than maximum likelihood estimation. This modification improves DRIP's statistical power, enabling us to increase the number of identified spectrum at a 1% false discovery rate on yeast and worm data sets.