 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInvariant Causal Set Covering Machines
Jun 07, 2023
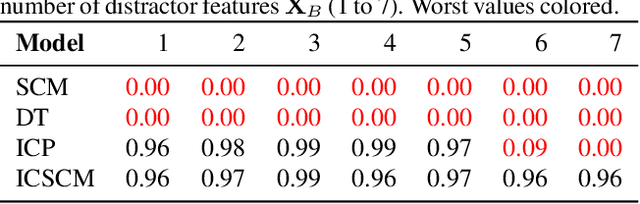

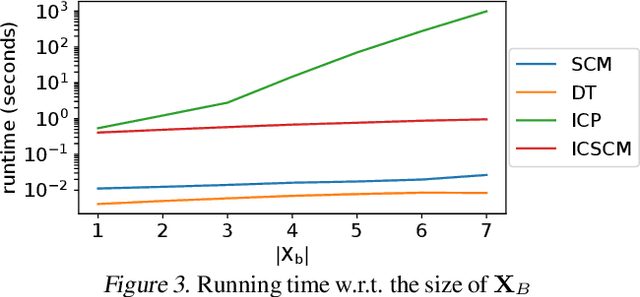
Rule-based models, such as decision trees, appeal to practitioners due to their interpretable nature. However, the learning algorithms that produce such models are often vulnerable to spurious associations and thus, they are not guaranteed to extract causally-relevant insights. In this work, we build on ideas from the invariant causal prediction literature to propose Invariant Causal Set Covering Machines, an extension of the classical Set Covering Machine algorithm for conjunctions/disjunctions of binary-valued rules that provably avoids spurious associations. We demonstrate both theoretically and empirically that our method can identify the causal parents of a variable of interest in polynomial time.
RandomSCM: interpretable ensembles of sparse classifiers tailored for omics data
Aug 11, 2022

Background: Understanding the relationship between the Omics and the phenotype is a central problem in precision medicine. The high dimensionality of metabolomics data challenges learning algorithms in terms of scalability and generalization. Most learning algorithms do not produce interpretable models -- Method: We propose an ensemble learning algorithm based on conjunctions or disjunctions of decision rules. -- Results : Applications on metabolomics data shows that it produces models that achieves high predictive performances. The interpretability of the models makes them useful for biomarker discovery and patterns discovery in high dimensional data.
Large scale modeling of antimicrobial resistance with interpretable classifiers
Dec 03, 2016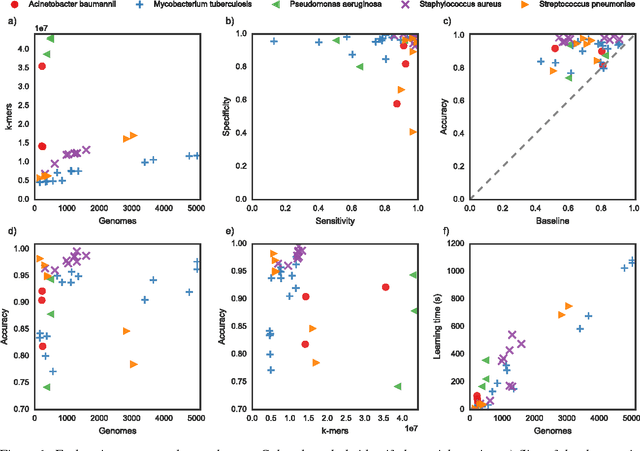
Antimicrobial resistance is an important public health concern that has implications in the practice of medicine worldwide. Accurately predicting resistance phenotypes from genome sequences shows great promise in promoting better use of antimicrobial agents, by determining which antibiotics are likely to be effective in specific clinical cases. In healthcare, this would allow for the design of treatment plans tailored for specific individuals, likely resulting in better clinical outcomes for patients with bacterial infections. In this work, we present the recent work of Drouin et al. (2016) on using Set Covering Machines to learn highly interpretable models of antibiotic resistance and complement it by providing a large scale application of their method to the entire PATRIC database. We report prediction results for 36 new datasets and present the Kover AMR platform, a new web-based tool allowing the visualization and interpretation of the generated models.
Greedy Biomarker Discovery in the Genome with Applications to Antimicrobial Resistance
May 22, 2015
The Set Covering Machine (SCM) is a greedy learning algorithm that produces sparse classifiers. We extend the SCM for datasets that contain a huge number of features. The whole genetic material of living organisms is an example of such a case, where the number of feature exceeds 10^7. Three human pathogens were used to evaluate the performance of the SCM at predicting antimicrobial resistance. Our results show that the SCM compares favorably in terms of sparsity and accuracy against L1 and L2 regularized Support Vector Machines and CART decision trees. Moreover, the SCM was the only algorithm that could consider the full feature space. For all other algorithms, the latter had to be filtered as a preprocessing step.
Learning interpretable models of phenotypes from whole genome sequences with the Set Covering Machine
Dec 02, 2014


The increased affordability of whole genome sequencing has motivated its use for phenotypic studies. We address the problem of learning interpretable models for discrete phenotypes from whole genomes. We propose a general approach that relies on the Set Covering Machine and a k-mer representation of the genomes. We show results for the problem of predicting the resistance of Pseudomonas Aeruginosa, an important human pathogen, against 4 antibiotics. Our results demonstrate that extremely sparse models which are biologically relevant can be learnt using this approach.
Learning a peptide-protein binding affinity predictor with kernel ridge regression
Jul 31, 2012We propose a specialized string kernel for small bio-molecules, peptides and pseudo-sequences of binding interfaces. The kernel incorporates physico-chemical properties of amino acids and elegantly generalize eight kernels, such as the Oligo, the Weighted Degree, the Blended Spectrum, and the Radial Basis Function. We provide a low complexity dynamic programming algorithm for the exact computation of the kernel and a linear time algorithm for it's approximation. Combined with kernel ridge regression and SupCK, a novel binding pocket kernel, the proposed kernel yields biologically relevant and good prediction accuracy on the PepX database. For the first time, a machine learning predictor is capable of accurately predicting the binding affinity of any peptide to any protein. The method was also applied to both single-target and pan-specific Major Histocompatibility Complex class II benchmark datasets and three Quantitative Structure Affinity Model benchmark datasets. On all benchmarks, our method significantly (p-value < 0.057) outperforms the current state-of-the-art methods at predicting peptide-protein binding affinities. The proposed approach is flexible and can be applied to predict any quantitative biological activity. The method should be of value to a large segment of the research community with the potential to accelerate peptide-based drug and vaccine development.
* 22 pages, 4 figures, 5 tables
Feature Selection with Conjunctions of Decision Stumps and Learning from Microarray Data
May 04, 2010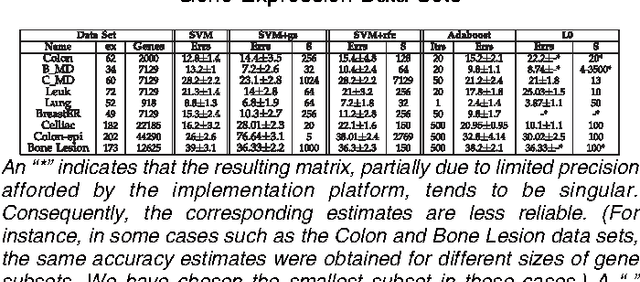



One of the objectives of designing feature selection learning algorithms is to obtain classifiers that depend on a small number of attributes and have verifiable future performance guarantees. There are few, if any, approaches that successfully address the two goals simultaneously. Performance guarantees become crucial for tasks such as microarray data analysis due to very small sample sizes resulting in limited empirical evaluation. To the best of our knowledge, such algorithms that give theoretical bounds on the future performance have not been proposed so far in the context of the classification of gene expression data. In this work, we investigate the premise of learning a conjunction (or disjunction) of decision stumps in Occam's Razor, Sample Compression, and PAC-Bayes learning settings for identifying a small subset of attributes that can be used to perform reliable classification tasks. We apply the proposed approaches for gene identification from DNA microarray data and compare our results to those of well known successful approaches proposed for the task. We show that our algorithm not only finds hypotheses with much smaller number of genes while giving competitive classification accuracy but also have tight risk guarantees on future performance unlike other approaches. The proposed approaches are general and extensible in terms of both designing novel algorithms and application to other domains.