 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBrain Mapping with Dense Features: Grounding Cortical Semantic Selectivity in Natural Images With Vision Transformers
Oct 07, 2024



Advances in large-scale artificial neural networks have facilitated novel insights into the functional topology of the brain. Here, we leverage this approach to study how semantic categories are organized in the human visual cortex. To overcome the challenge presented by the co-occurrence of multiple categories in natural images, we introduce BrainSAIL (Semantic Attribution and Image Localization), a method for isolating specific neurally-activating visual concepts in images. BrainSAIL exploits semantically consistent, dense spatial features from pre-trained vision models, building upon their demonstrated ability to robustly predict neural activity. This method derives clean, spatially dense embeddings without requiring any additional training, and employs a novel denoising process that leverages the semantic consistency of images under random augmentations. By unifying the space of whole-image embeddings and dense visual features and then applying voxel-wise encoding models to these features, we enable the identification of specific subregions of each image which drive selectivity patterns in different areas of the higher visual cortex. We validate BrainSAIL on cortical regions with known category selectivity, demonstrating its ability to accurately localize and disentangle selectivity to diverse visual concepts. Next, we demonstrate BrainSAIL's ability to characterize high-level visual selectivity to scene properties and low-level visual features such as depth, luminance, and saturation, providing insights into the encoding of complex visual information. Finally, we use BrainSAIL to directly compare the feature selectivity of different brain encoding models across different regions of interest in visual cortex. Our innovative method paves the way for significant advances in mapping and decomposing high-level visual representations in the human brain.
Neural Representations of Dynamic Visual Stimuli
Jun 04, 2024



Humans experience the world through constantly changing visual stimuli, where scenes can shift and move, change in appearance, and vary in distance. The dynamic nature of visual perception is a fundamental aspect of our daily lives, yet the large majority of research on object and scene processing, particularly using fMRI, has focused on static stimuli. While studies of static image perception are attractive due to their computational simplicity, they impose a strong non-naturalistic constraint on our investigation of human vision. In contrast, dynamic visual stimuli offer a more ecologically-valid approach but present new challenges due to the interplay between spatial and temporal information, making it difficult to disentangle the representations of stable image features and motion. To overcome this limitation -- given dynamic inputs, we explicitly decouple the modeling of static image representations and motion representations in the human brain. Three results demonstrate the feasibility of this approach. First, we show that visual motion information as optical flow can be predicted (or decoded) from brain activity as measured by fMRI. Second, we show that this predicted motion can be used to realistically animate static images using a motion-conditioned video diffusion model (where the motion is driven by fMRI brain activity). Third, we show prediction in the reverse direction: existing video encoders can be fine-tuned to predict fMRI brain activity from video imagery, and can do so more effectively than image encoders. This foundational work offers a novel, extensible framework for interpreting how the human brain processes dynamic visual information.
DeepChrome 2.0: Investigating and Improving Architectures, Visualizations, & Experiments
Sep 24, 2022
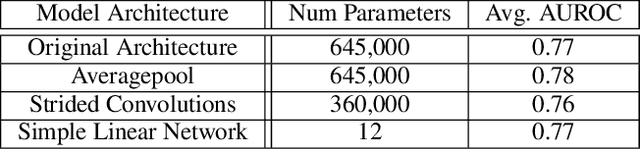
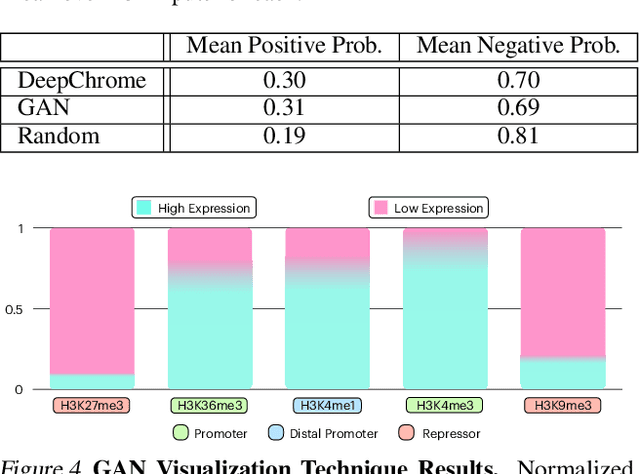
Histone modifications play a critical role in gene regulation. Consequently, predicting gene expression from histone modification signals is a highly motivated problem in epigenetics. We build upon the work of DeepChrome by Singh et al. (2016), who trained classifiers that map histone modification signals to gene expression. We present a novel visualization technique for providing insight into combinatorial relationships among histone modifications for gene regulation that uses a generative adversarial network to generate histone modification signals. We also explore and compare various architectural changes, with results suggesting that the 645k-parameter convolutional neural network from DeepChrome has the same predictive power as a 12-parameter linear network. Results from cross-cell prediction experiments, where the model is trained and tested on datasets of varying sizes, cell-types, and correlations, suggest the relationship between histone modification signals and gene expression is independent of cell type. We release our PyTorch re-implementation of DeepChrome on GitHub \footnote{\url{github.com/ssss1029/gene_expression_294}}.\parfillskip=0pt