 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOrb: A Fast, Scalable Neural Network Potential
Oct 29, 2024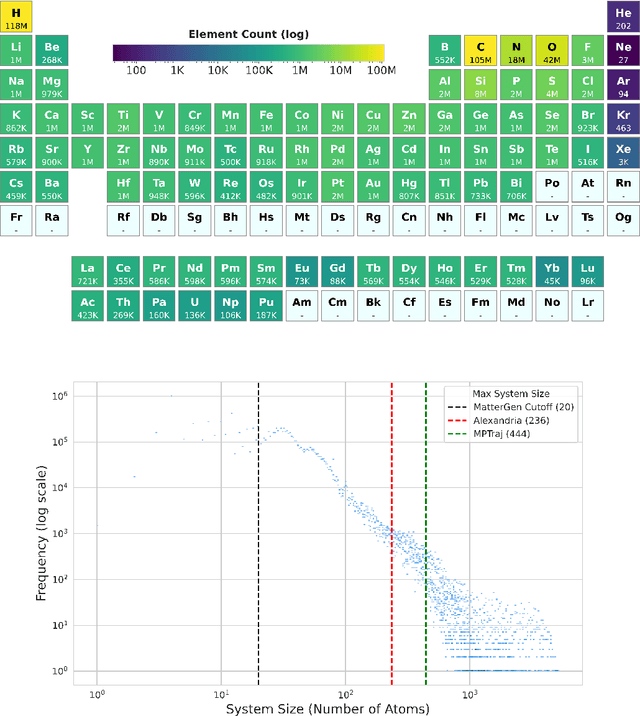
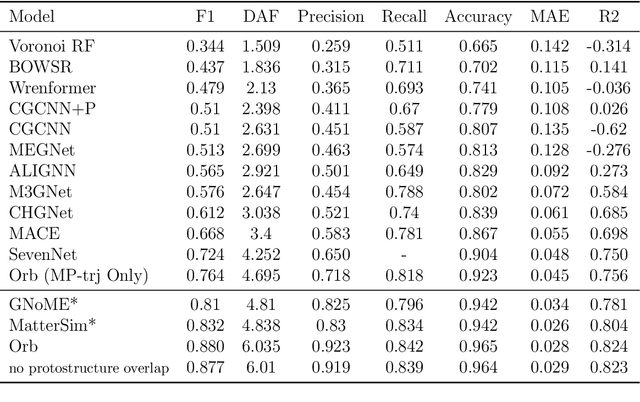
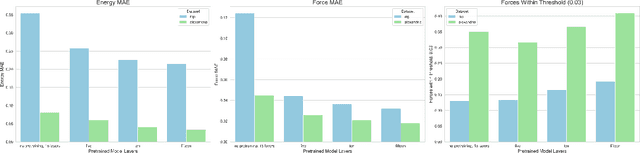
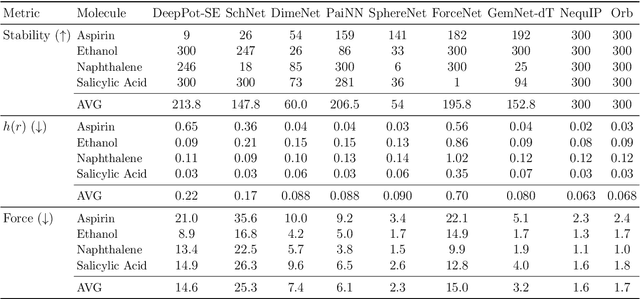
We introduce Orb, a family of universal interatomic potentials for atomistic modelling of materials. Orb models are 3-6 times faster than existing universal potentials, stable under simulation for a range of out of distribution materials and, upon release, represented a 31% reduction in error over other methods on the Matbench Discovery benchmark. We explore several aspects of foundation model development for materials, with a focus on diffusion pretraining. We evaluate Orb as a model for geometry optimization, Monte Carlo and molecular dynamics simulations.
Closed-loop Error Correction Learning Accelerates Experimental Discovery of Thermoelectric Materials
Feb 26, 2023The exploration of thermoelectric materials is challenging considering the large materials space, combined with added exponential degrees of freedom coming from doping and the diversity of synthetic pathways. Here we seek to incorporate historical data and update and refine it using experimental feedback by employing error-correction learning (ECL). We thus learn from prior datasets and then adapt the model to differences in synthesis and characterization that are otherwise difficult to parameterize. We then apply this strategy to discovering thermoelectric materials where we prioritize synthesis at temperatures < 300{\deg}C. We document a previously unreported chemical family of thermoelectric materials, PbSe:SnSb, finding that the best candidate in this chemical family, 2 wt% SnSb doped PbSe, exhibits a power factor more than 2x that of PbSe. Our investigations show that our closed-loop experimentation strategy reduces the required number of experiments to find an optimized material by as much as 3x compared to high-throughput searches powered by state-of-the-art machine learning models. We also observe that this improvement is dependent on the accuracy of prior in a manner that exhibits diminishing returns, and after a certain accuracy is reached, it is factors associated with experimental pathways that dictate the trends.
Interpretable discovery of new semiconductors with machine learning
Jan 12, 2021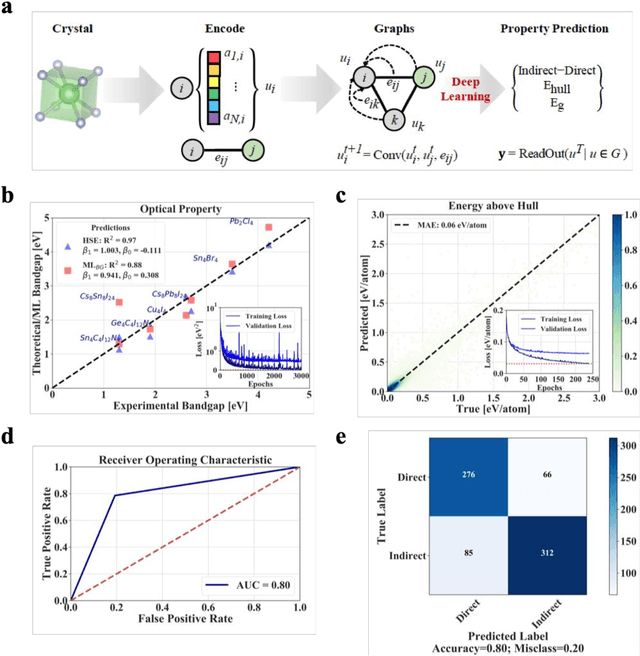
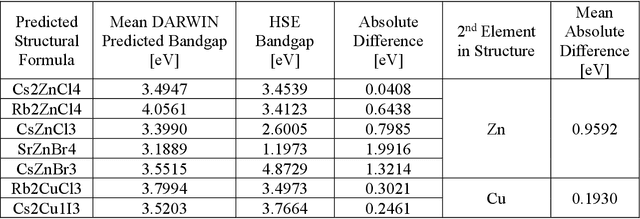
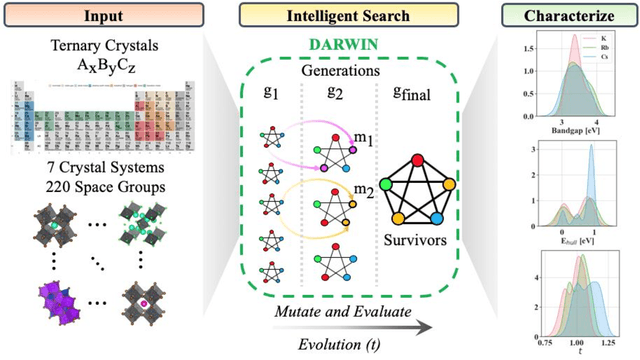
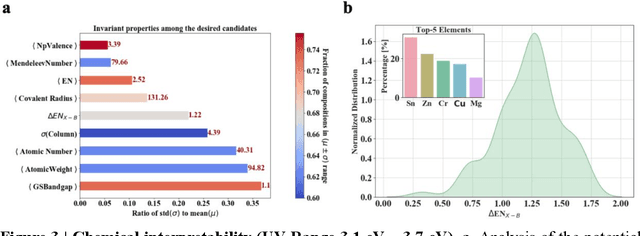
Machine learning models of materials$^{1-5}$ accelerate discovery compared to ab initio methods: deep learning models now reproduce density functional theory (DFT)-calculated results at one hundred thousandths of the cost of DFT$^{6}$. To provide guidance in experimental materials synthesis, these need to be coupled with an accurate yet effective search algorithm and training data consistent with experimental observations. Here we report an evolutionary algorithm powered search which uses machine-learned surrogate models trained on high-throughput hybrid functional DFT data benchmarked against experimental bandgaps: Deep Adaptive Regressive Weighted Intelligent Network (DARWIN). The strategy enables efficient search over the materials space of ~10$^8$ ternaries and 10$^{11}$ quaternaries$^{7}$ for candidates with target properties. It provides interpretable design rules, such as our finding that the difference in the electronegativity between the halide and B-site cation being a strong predictor of ternary structural stability. As an example, when we seek UV emission, DARWIN predicts K$_2$CuX$_3$ (X = Cl, Br) as a promising materials family, based on its electronegativity difference. We synthesized and found these materials to be stable, direct bandgap UV emitters. The approach also allows knowledge distillation for use by humans.