 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInspect, Understand, Overcome: A Survey of Practical Methods for AI Safety
Apr 29, 2021The use of deep neural networks (DNNs) in safety-critical applications like mobile health and autonomous driving is challenging due to numerous model-inherent shortcomings. These shortcomings are diverse and range from a lack of generalization over insufficient interpretability to problems with malicious inputs. Cyber-physical systems employing DNNs are therefore likely to suffer from safety concerns. In recent years, a zoo of state-of-the-art techniques aiming to address these safety concerns has emerged. This work provides a structured and broad overview of them. We first identify categories of insufficiencies to then describe research activities aiming at their detection, quantification, or mitigation. Our paper addresses both machine learning experts and safety engineers: The former ones might profit from the broad range of machine learning topics covered and discussions on limitations of recent methods. The latter ones might gain insights into the specifics of modern ML methods. We moreover hope that our contribution fuels discussions on desiderata for ML systems and strategies on how to propel existing approaches accordingly.
By-passing the Kohn-Sham equations with machine learning
Jun 15, 2017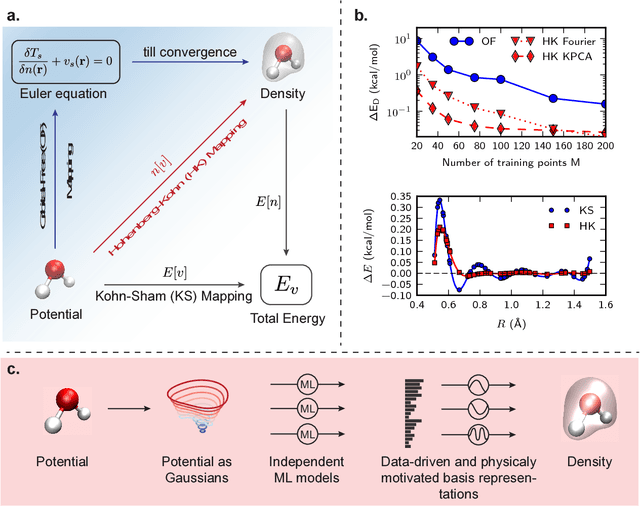
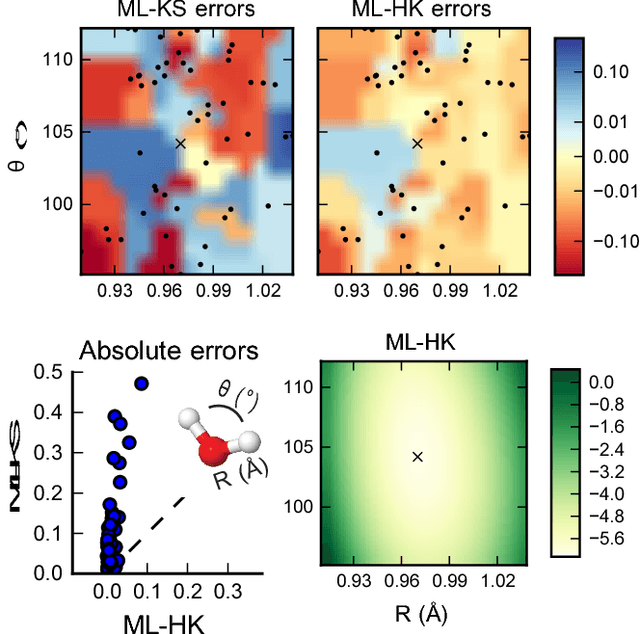
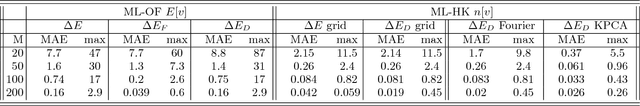

Last year, at least 30,000 scientific papers used the Kohn-Sham scheme of density functional theory to solve electronic structure problems in a wide variety of scientific fields, ranging from materials science to biochemistry to astrophysics. Machine learning holds the promise of learning the kinetic energy functional via examples, by-passing the need to solve the Kohn-Sham equations. This should yield substantial savings in computer time, allowing either larger systems or longer time-scales to be tackled, but attempts to machine-learn this functional have been limited by the need to find its derivative. The present work overcomes this difficulty by directly learning the density-potential and energy-density maps for test systems and various molecules. Both improved accuracy and lower computational cost with this method are demonstrated by reproducing DFT energies for a range of molecular geometries generated during molecular dynamics simulations. Moreover, the methodology could be applied directly to quantum chemical calculations, allowing construction of density functionals of quantum-chemical accuracy.