 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnhancing molecular dynamics with equivariant machine-learned densities
Apr 27, 2026Machine-learning interatomic potentials (MLIPs) have enabled molecular dynamics at near ab initio accuracy, yet remain limited to energies and forces by construction, leaving electronic observables such as dipole moments and polarizabilities inaccessible. We introduce DenSNet, a density-first approach to machine-learned electronic structure that learns the Hohenberg--Kohn map from nuclear configurations to the ground-state electron density. Our approach employs an SE(3)-equivariant neural network to predict density coefficients of a flexible atom-centered Gaussian basis, combined with a $Δ$-learning strategy that uses superposed atomic densities as a prior to accelerate training. A second equivariant network then maps the predicted density to the total energy, providing a unified framework for molecular dynamics and electronic structure. We validate DenSNet on ethanol, ethanethiol, and resorcinol, where infrared spectra from machine-learned trajectories show excellent agreement with experimental gas-phase measurements. To test scalability, we train on polythiophene oligomers with 1--6 monomers and extrapolate to chains of up to 12 monomers, generating stable long-time trajectories whose infrared spectra agree with reference density functional theory calculations. Here, we show that reinstating the electron density as the central learned quantity opens a practical route to transferable prediction of spectroscopic and electronic observables in large-scale molecular simulations.
MolCryst-MLIPs: A Machine-Learned Interatomic Potentials Database for Molecular Crystals
Apr 15, 2026We present an open Molecular Crystal (MC) database of Machine-Learned Interatomic Potentials (MLIP) called MolCryst-MLIPs. The first release comprises fine-tuned MACE models for nine molecular crystal systems -- Benzamide, Benzoic acid, Coumarin, Durene, Isonicotinamide, Niacinamide, Nicotinamide, Pyrazinamide, and Resorcinol -- developed using the Automated Machine Learning Pipeline (AMLP), which streamlines the entire MLIP development workflow, from reference data generation to model training and validation, into a reproducible and user-friendly pipeline. Models are fine-tuned from the MACE-MH-1 foundation model (omol head), yielding a mean energy MAE of 0.141 kJ/mol/atom and a mean force MAE of 0.648 kJ/mol/Angstrom across all systems. Dynamical stability and structural integrity, as assessed through energy conservation, P2 orientational order parameters, and radial distribution functions, are evaluated using molecular dynamics simulations. The released models and datasets constitute a growing open database of validated MLIPs, ready for production MD simulations of molecular crystal polymorphism under different thermodynamic conditions.
On the design space between molecular mechanics and machine learning force fields
Sep 03, 2024A force field as accurate as quantum mechanics (QM) and as fast as molecular mechanics (MM), with which one can simulate a biomolecular system efficiently enough and meaningfully enough to get quantitative insights, is among the most ardent dreams of biophysicists -- a dream, nevertheless, not to be fulfilled any time soon. Machine learning force fields (MLFFs) represent a meaningful endeavor towards this direction, where differentiable neural functions are parametrized to fit ab initio energies, and furthermore forces through automatic differentiation. We argue that, as of now, the utility of the MLFF models is no longer bottlenecked by accuracy but primarily by their speed (as well as stability and generalizability), as many recent variants, on limited chemical spaces, have long surpassed the chemical accuracy of $1$ kcal/mol -- the empirical threshold beyond which realistic chemical predictions are possible -- though still magnitudes slower than MM. Hoping to kindle explorations and designs of faster, albeit perhaps slightly less accurate MLFFs, in this review, we focus our attention on the design space (the speed-accuracy tradeoff) between MM and ML force fields. After a brief review of the building blocks of force fields of either kind, we discuss the desired properties and challenges now faced by the force field development community, survey the efforts to make MM force fields more accurate and ML force fields faster, envision what the next generation of MLFF might look like.
By-passing the Kohn-Sham equations with machine learning
Jun 15, 2017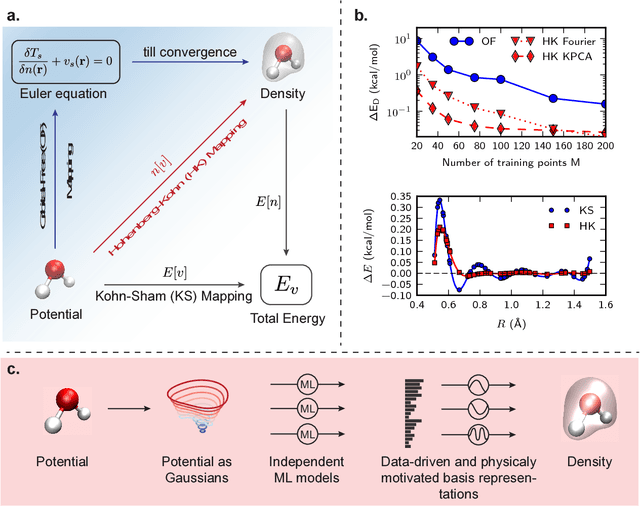
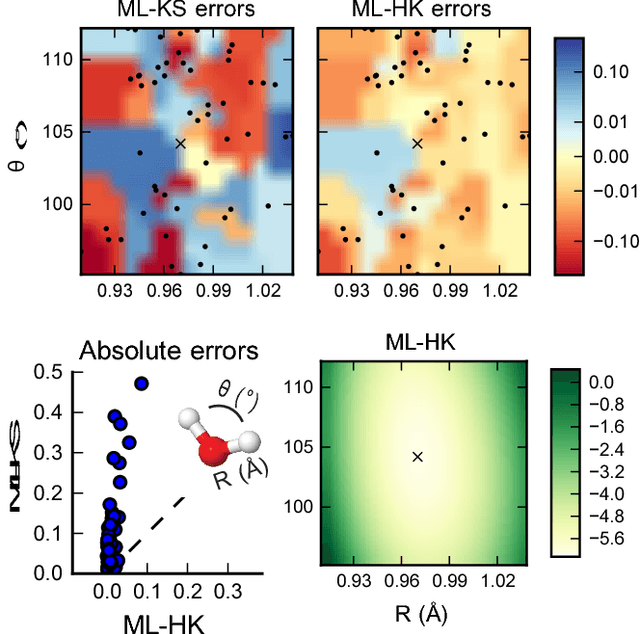
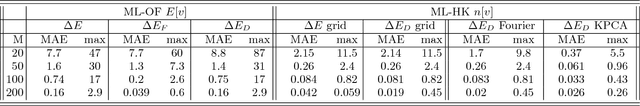

Last year, at least 30,000 scientific papers used the Kohn-Sham scheme of density functional theory to solve electronic structure problems in a wide variety of scientific fields, ranging from materials science to biochemistry to astrophysics. Machine learning holds the promise of learning the kinetic energy functional via examples, by-passing the need to solve the Kohn-Sham equations. This should yield substantial savings in computer time, allowing either larger systems or longer time-scales to be tackled, but attempts to machine-learn this functional have been limited by the need to find its derivative. The present work overcomes this difficulty by directly learning the density-potential and energy-density maps for test systems and various molecules. Both improved accuracy and lower computational cost with this method are demonstrated by reproducing DFT energies for a range of molecular geometries generated during molecular dynamics simulations. Moreover, the methodology could be applied directly to quantum chemical calculations, allowing construction of density functionals of quantum-chemical accuracy.