 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePathologist-like explainable AI for interpretable Gleason grading in prostate cancer
Oct 19, 2024



The aggressiveness of prostate cancer, the most common cancer in men worldwide, is primarily assessed based on histopathological data using the Gleason scoring system. While artificial intelligence (AI) has shown promise in accurately predicting Gleason scores, these predictions often lack inherent explainability, potentially leading to distrust in human-machine interactions. To address this issue, we introduce a novel dataset of 1,015 tissue microarray core images, annotated by an international group of 54 pathologists. The annotations provide detailed localized pattern descriptions for Gleason grading in line with international guidelines. Utilizing this dataset, we develop an inherently explainable AI system based on a U-Net architecture that provides predictions leveraging pathologists' terminology. This approach circumvents post-hoc explainability methods while maintaining or exceeding the performance of methods trained directly for Gleason pattern segmentation (Dice score: 0.713 $\pm$ 0.003 trained on explanations vs. 0.691 $\pm$ 0.010 trained on Gleason patterns). By employing soft labels during training, we capture the intrinsic uncertainty in the data, yielding strong results in Gleason pattern segmentation even in the context of high interobserver variability. With the release of this dataset, we aim to encourage further research into segmentation in medical tasks with high levels of subjectivity and to advance the understanding of pathologists' reasoning processes.
Deep Hypothesis Tests Detect Clinically Relevant Subgroup Shifts in Medical Images
Mar 08, 2023



Distribution shifts remain a fundamental problem for the safe application of machine learning systems. If undetected, they may impact the real-world performance of such systems or will at least render original performance claims invalid. In this paper, we focus on the detection of subgroup shifts, a type of distribution shift that can occur when subgroups have a different prevalence during validation compared to the deployment setting. For example, algorithms developed on data from various acquisition settings may be predominantly applied in hospitals with lower quality data acquisition, leading to an inadvertent performance drop. We formulate subgroup shift detection in the framework of statistical hypothesis testing and show that recent state-of-the-art statistical tests can be effectively applied to subgroup shift detection on medical imaging data. We provide synthetic experiments as well as extensive evaluation on clinically meaningful subgroup shifts on histopathology as well as retinal fundus images. We conclude that classifier-based subgroup shift detection tests could be a particularly useful tool for post-market surveillance of deployed ML systems.
Studying Therapy Effects and Disease Outcomes in Silico using Artificial Counterfactual Tissue Samples
Feb 06, 2023
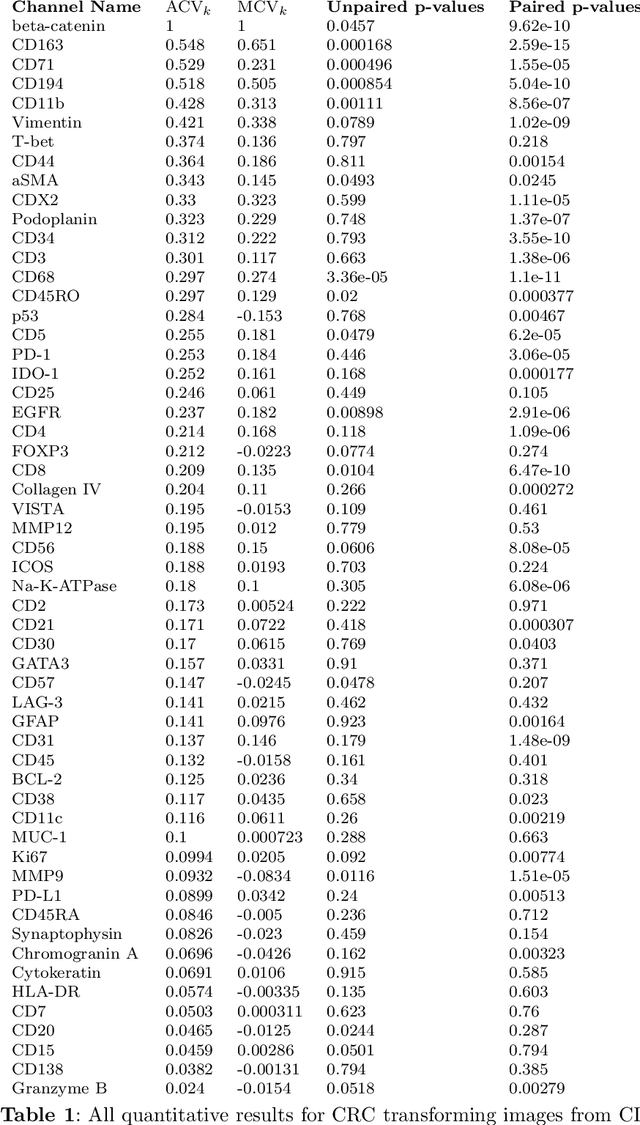
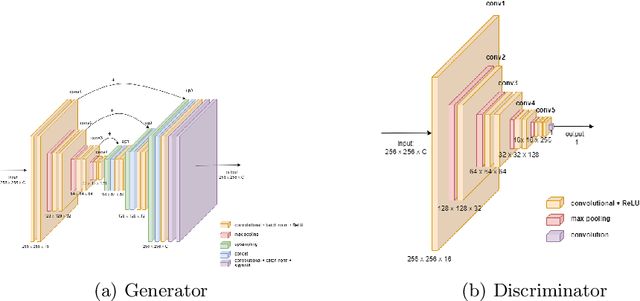
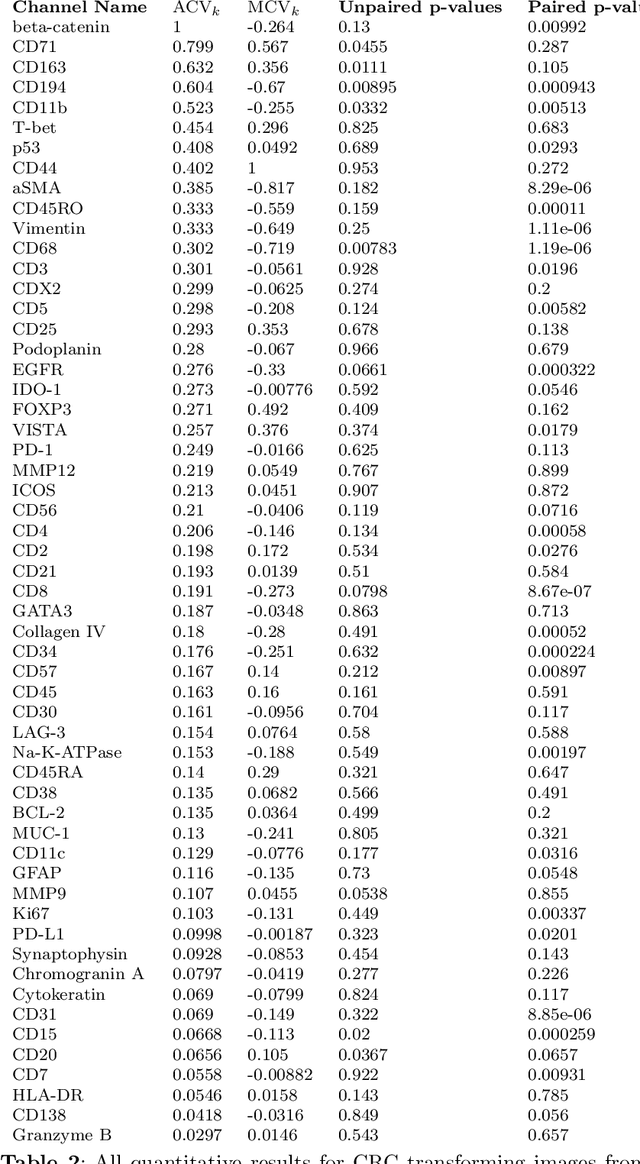
Understanding the interactions of different cell types inside the immune tumor microenvironment (iTME) is crucial for the development of immunotherapy treatments as well as for predicting their outcomes. Highly multiplexed tissue imaging (HMTI) technologies offer a tool which can capture cell properties of tissue samples by measuring expression of various proteins and storing them in separate image channels. HMTI technologies can be used to gain insights into the iTME and in particular how the iTME differs for different patient outcome groups of interest (e.g., treatment responders vs. non-responders). Understanding the systematic differences in the iTME of different patient outcome groups is crucial for developing better treatments and personalising existing treatments. However, such analyses are inherently limited by the fact that any two tissue samples vary due to a large number of factors unrelated to the outcome. Here, we present CF-HistoGAN, a machine learning framework that employs generative adversarial networks (GANs) to create artificial counterfactual tissue samples that resemble the original tissue samples as closely as possible but capture the characteristics of a different patient outcome group. Specifically, we learn to "translate" HMTI samples from one patient group to create artificial paired samples. We show that this approach allows to directly study the effects of different patient outcomes on the iTMEs of individual tissue samples. We demonstrate that CF-HistoGAN can be employed as an explorative tool for understanding iTME effects on the pixel level. Moreover, we show that our method can be used to identify statistically significant differences in the expression of different proteins between patient groups with greater sensitivity compared to conventional approaches.