 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning To Navigate The Synthetically Accessible Chemical Space Using Reinforcement Learning
May 20, 2020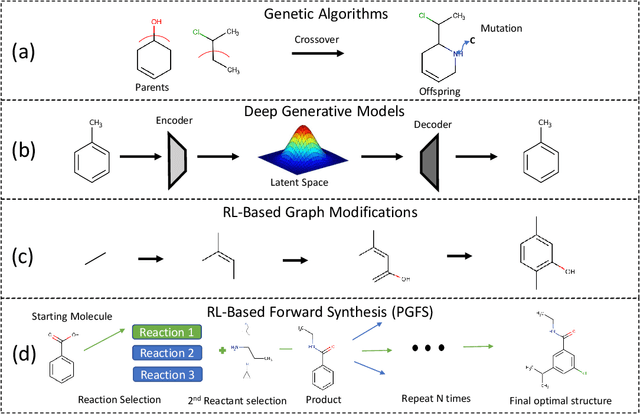
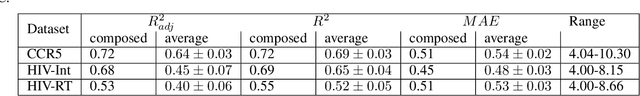

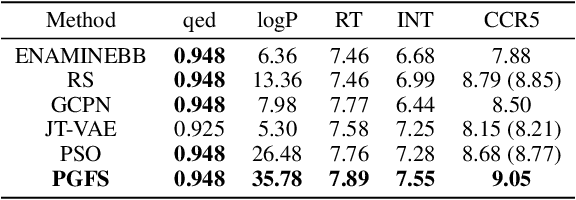
Over the last decade, there has been significant progress in the field of machine learning for de novo drug design, particularly in deep generative models. However, current generative approaches exhibit a significant challenge as they do not ensure that the proposed molecular structures can be feasibly synthesized nor do they provide the synthesis routes of the proposed small molecules, thereby seriously limiting their practical applicability. In this work, we propose a novel forward synthesis framework powered by reinforcement learning (RL) for de novo drug design, Policy Gradient for Forward Synthesis (PGFS), that addresses this challenge by embedding the concept of synthetic accessibility directly into the de novo drug design system. In this setup, the agent learns to navigate through the immense synthetically accessible chemical space by subjecting commercially available small molecule building blocks to valid chemical reactions at every time step of the iterative virtual multi-step synthesis process. The proposed environment for drug discovery provides a highly challenging test-bed for RL algorithms owing to the large state space and high-dimensional continuous action space with hierarchical actions. PGFS achieves state-of-the-art performance in generating structures with high QED and penalized clogP. Moreover, we validate PGFS in an in-silico proof-of-concept associated with three HIV targets. Finally, we describe how the end-to-end training conceptualized in this study represents an important paradigm in radically expanding the synthesizable chemical space and automating the drug discovery process.
Improving Chemical Autoencoder Latent Space and Molecular De novo Generation Diversity with Heteroencoders
Sep 17, 2018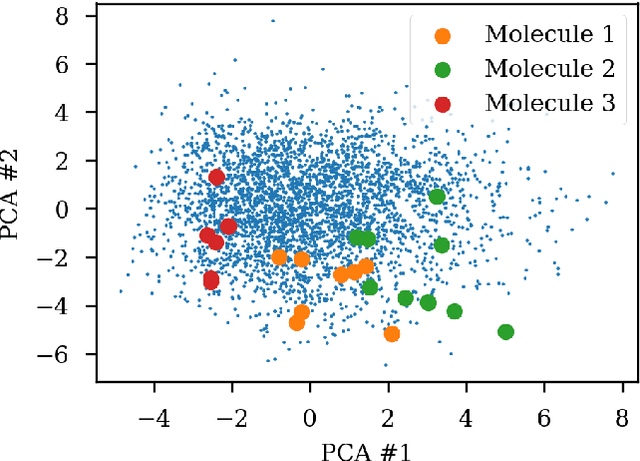
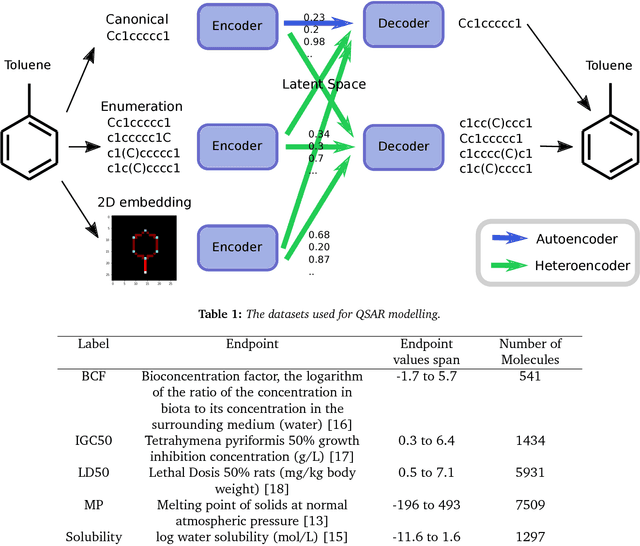

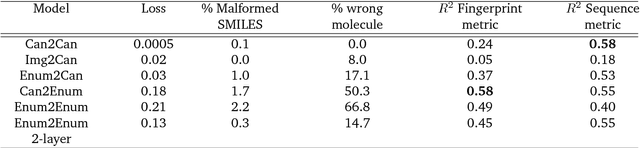
Chemical autoencoders are attractive models as they combine chemical space navigation with possibilities for de-novo molecule generation in areas of interest. This enables them to produce focused chemical libraries around a single lead compound for employment early in a drug discovery project. Here it is shown that the choice of chemical representation, such as SMILES strings, has a large influence on the properties of the latent space. It is further explored to what extent translating between different chemical representations influences the latent space similarity to the SMILES strings or circular fingerprints. By employing SMILES enumeration for either the encoder or decoder, it is found that the decoder has the largest influence on the properties of the latent space. Training a sequence to sequence heteroencoder based on recurrent neural networks(RNNs) with long short-term memory cells (LSTM) to predict different enumerated SMILES strings from the same canonical SMILES string gives the largest similarity between latent space distance and molecular similarity measured as circular fingerprints similarity. Using the output from the bottleneck in QSAR modelling of five molecular datasets shows that heteroencoder derived vectors markedly outperforms autoencoder derived vectors as well as models built using ECFP4 fingerprints, underlining the increased chemical relevance of the latent space. However, the use of enumeration during training of the decoder leads to a markedly increase in the rate of decoding to a different molecules than encoded, a tendency that can be counteracted with more complex network architectures.