 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving Chemical Autoencoder Latent Space and Molecular De novo Generation Diversity with Heteroencoders
Sep 17, 2018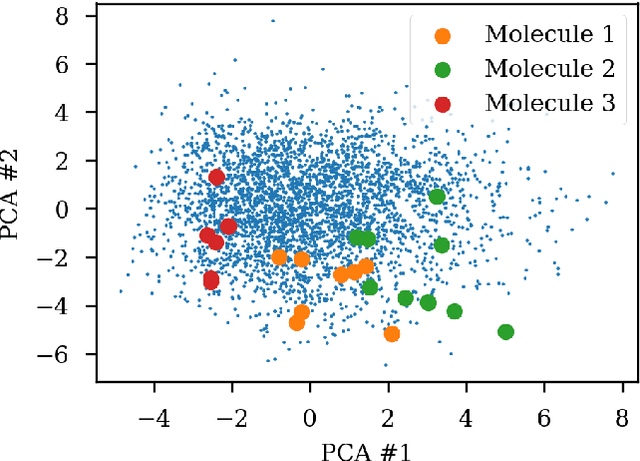
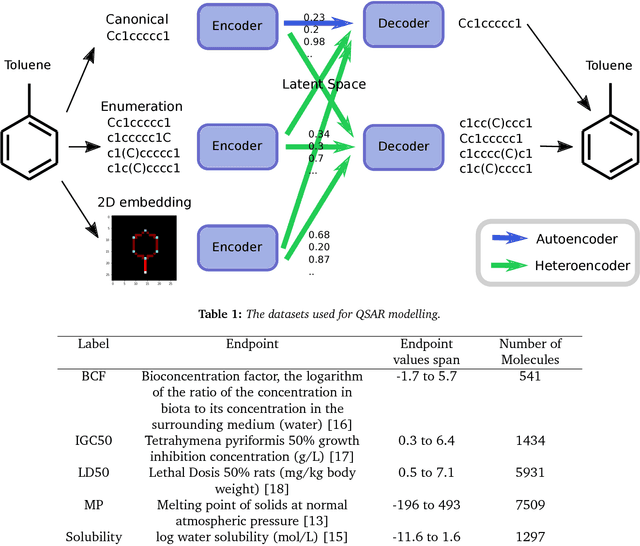

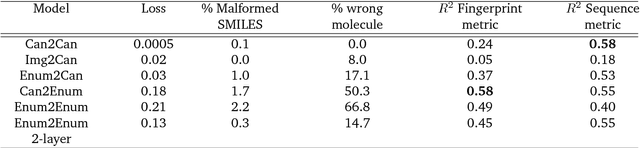
Chemical autoencoders are attractive models as they combine chemical space navigation with possibilities for de-novo molecule generation in areas of interest. This enables them to produce focused chemical libraries around a single lead compound for employment early in a drug discovery project. Here it is shown that the choice of chemical representation, such as SMILES strings, has a large influence on the properties of the latent space. It is further explored to what extent translating between different chemical representations influences the latent space similarity to the SMILES strings or circular fingerprints. By employing SMILES enumeration for either the encoder or decoder, it is found that the decoder has the largest influence on the properties of the latent space. Training a sequence to sequence heteroencoder based on recurrent neural networks(RNNs) with long short-term memory cells (LSTM) to predict different enumerated SMILES strings from the same canonical SMILES string gives the largest similarity between latent space distance and molecular similarity measured as circular fingerprints similarity. Using the output from the bottleneck in QSAR modelling of five molecular datasets shows that heteroencoder derived vectors markedly outperforms autoencoder derived vectors as well as models built using ECFP4 fingerprints, underlining the increased chemical relevance of the latent space. However, the use of enumeration during training of the decoder leads to a markedly increase in the rate of decoding to a different molecules than encoded, a tendency that can be counteracted with more complex network architectures.
DeepIEP: a Peptide Sequence Model of Isoelectric Point (IEP/pI) using Recurrent Neural Networks (RNNs)
Dec 27, 2017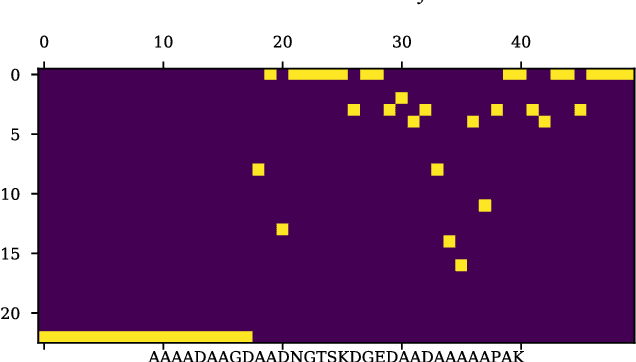
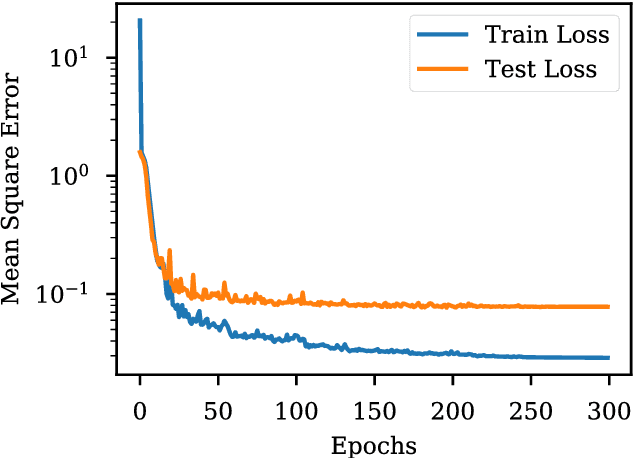
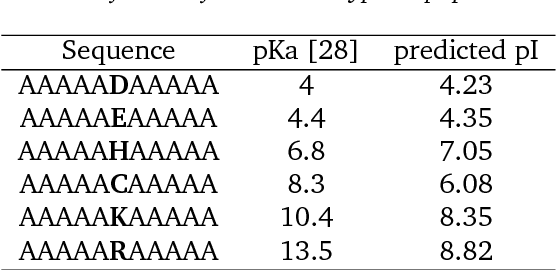
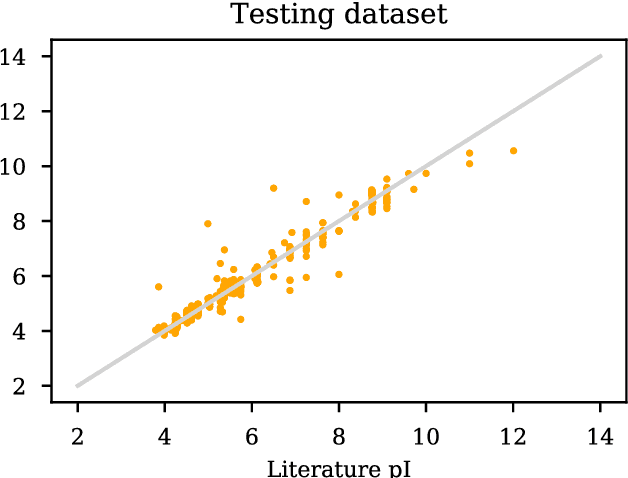
The isoelectric point (IEP or pI) is the pH where the net charge on the molecular ensemble of peptides and proteins is zero. This physical-chemical property is dependent on protonable/deprotonable sidechains and their pKa values. Here an pI prediction model is trained from a database of peptide sequences and pIs using a recurrent neural network (RNN) with long short-term memory (LSTM) cells. The trained model obtains an RMSE and R$^2$ of 0.28 and 0.95 for the external test set. The model is not based on pKa values, but prediction of constructed test sequences show similar rankings as already known pKa values. The prediction depends mostly on the existence of known acidic and basic amino acids with fine-adjusted based on the neighboring sequence and position of the charged amino acids in the peptide chain.
Data Augmentation of Spectral Data for Convolutional Neural Network (CNN) Based Deep Chemometrics
Oct 05, 2017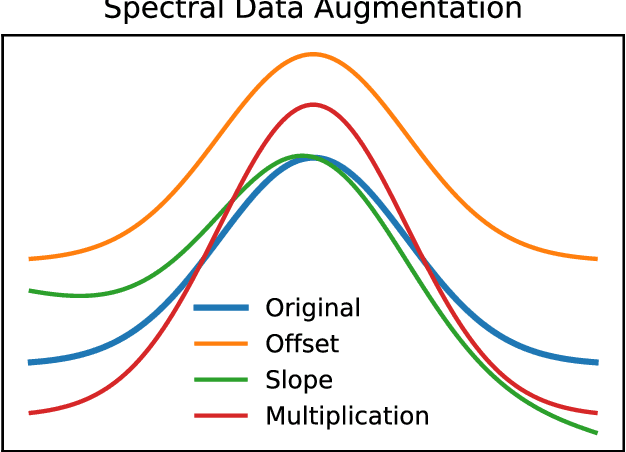
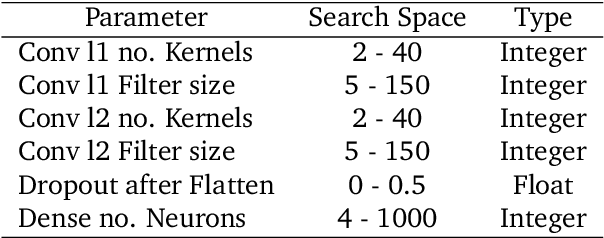
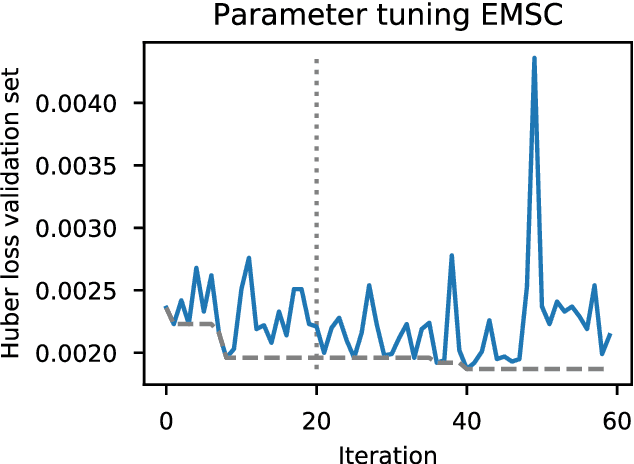
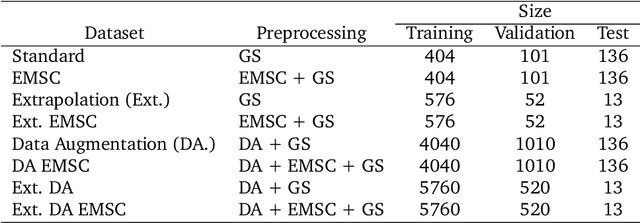
Deep learning methods are used on spectroscopic data to predict drug content in tablets from near infrared (NIR) spectra. Using convolutional neural networks (CNNs), features are ex- tracted from the spectroscopic data. Extended multiplicative scatter correction (EMSC) and a novel spectral data augmentation method are benchmarked as preprocessing steps. The learned models perform better or on par with hypothetical optimal partial least squares (PLS) models for all combinations of preprocessing. Data augmentation with subsequent EMSC in combination gave the best results. The deep learning model CNNs also outperform the PLS models in an extrapolation chal- lenge created using data from a second instrument and from an analyte concentration not covered by the training data. Qualitative investigations of the CNNs kernel activations show their resemblance to wellknown data processing methods such as smoothing, slope/derivative, thresholds and spectral region selection.
SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules
May 17, 2017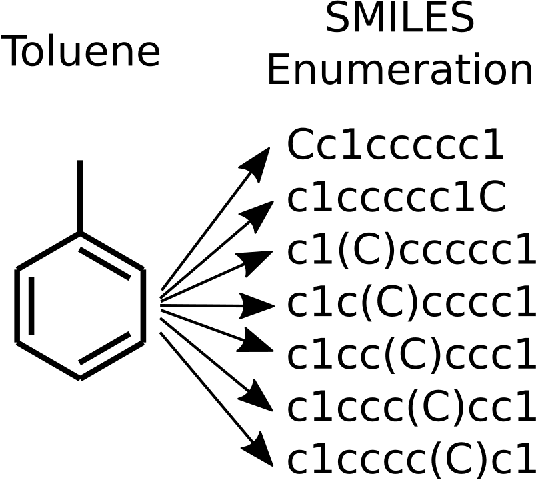
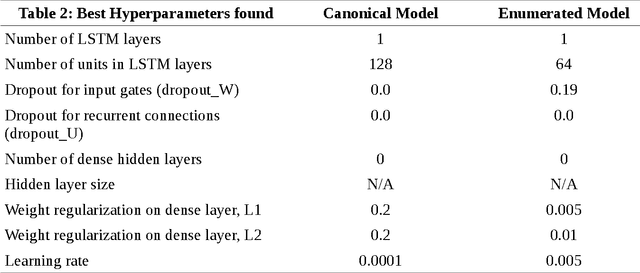
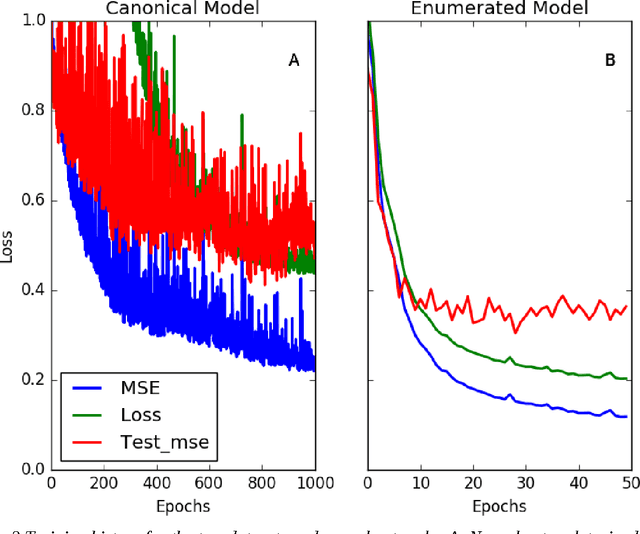
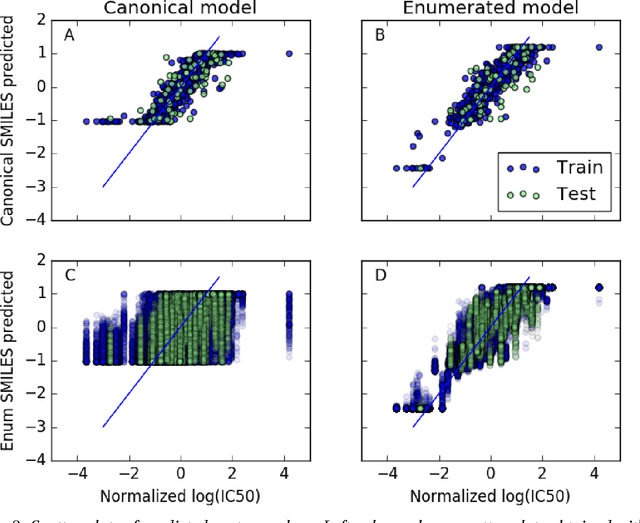
Simplified Molecular Input Line Entry System (SMILES) is a single line text representation of a unique molecule. One molecule can however have multiple SMILES strings, which is a reason that canonical SMILES have been defined, which ensures a one to one correspondence between SMILES string and molecule. Here the fact that multiple SMILES represent the same molecule is explored as a technique for data augmentation of a molecular QSAR dataset modeled by a long short term memory (LSTM) cell based neural network. The augmented dataset was 130 times bigger than the original. The network trained with the augmented dataset shows better performance on a test set when compared to a model built with only one canonical SMILES string per molecule. The correlation coefficient R2 on the test set was improved from 0.56 to 0.66 when using SMILES enumeration, and the root mean square error (RMS) likewise fell from 0.62 to 0.55. The technique also works in the prediction phase. By taking the average per molecule of the predictions for the enumerated SMILES a further improvement to a correlation coefficient of 0.68 and a RMS of 0.52 was found.
Molecular Generation with Recurrent Neural Networks (RNNs)
May 17, 2017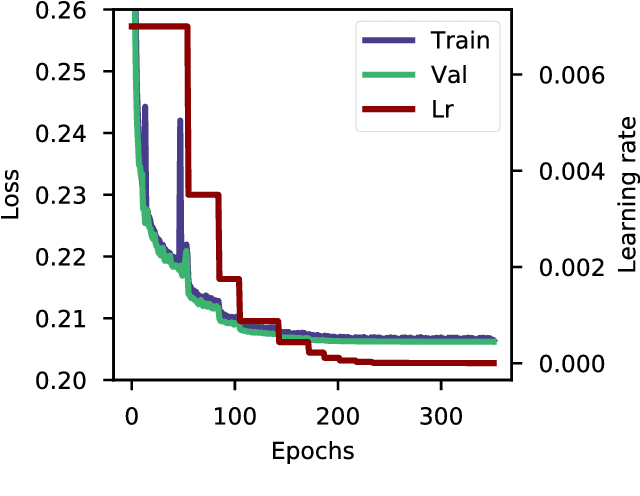

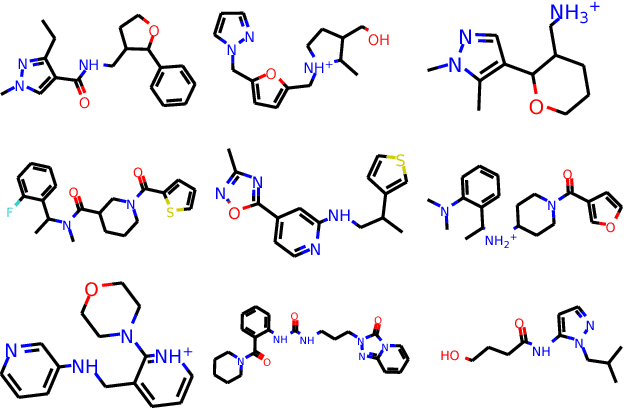
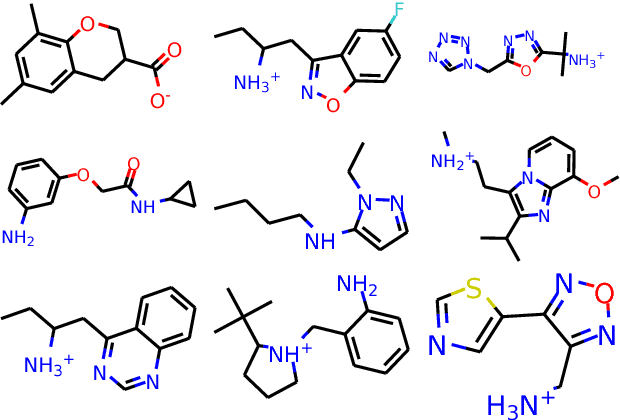
The potential number of drug like small molecules is estimated to be between 10^23 and 10^60 while current databases of known compounds are orders of magnitude smaller with approximately 10^8 compounds. This discrepancy has led to an interest in generating virtual libraries using hand crafted chemical rules and fragment based methods to cover a larger area of chemical space and generate chemical libraries for use in in silico drug discovery endeavors. Here it is explored to what extent a recurrent neural network with long short term memory cells can figure out sensible chemical rules and generate synthesizable molecules by being trained on existing compounds encoded as SMILES. The networks can to a high extent generate novel, but chemically sensible molecules. The properties of the molecules are tuned by training on two different datasets consisting of fragment like molecules and drug like molecules. The produced molecules and the training databases have very similar distributions of molar weight, predicted logP, number of hydrogen bond acceptors and donors, number of rotatable bonds and topological polar surface area when compared to their respective training sets. The compounds are for the most cases synthesizable as assessed with SA score and Wiley ChemPlanner.