 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReducing the Cost of Quantum Chemical Data By Backpropagating Through Density Functional Theory
Feb 06, 2024Density Functional Theory (DFT) accurately predicts the quantum chemical properties of molecules, but scales as $O(N_{\text{electrons}}^3)$. Sch\"utt et al. (2019) successfully approximate DFT 1000x faster with Neural Networks (NN). Arguably, the biggest problem one faces when scaling to larger molecules is the cost of DFT labels. For example, it took years to create the PCQ dataset (Nakata & Shimazaki, 2017) on which subsequent NNs are trained within a week. DFT labels molecules by minimizing energy $E(\cdot )$ as a "loss function." We bypass dataset creation by directly training NNs with $E(\cdot )$ as a loss function. For comparison, Sch\"utt et al. (2019) spent 626 hours creating a dataset on which they trained their NN for 160h, for a total of 786h; our method achieves comparable performance within 31h.
BESS: Balanced Entity Sampling and Sharing for Large-Scale Knowledge Graph Completion
Nov 22, 2022
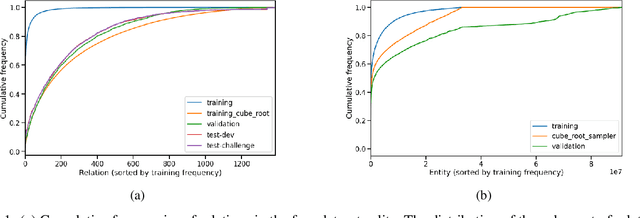

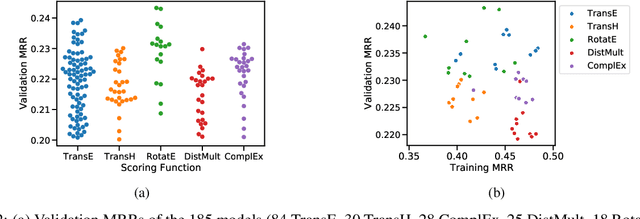
We present the award-winning submission to the WikiKG90Mv2 track of OGB-LSC@NeurIPS 2022. The task is link-prediction on the large-scale knowledge graph WikiKG90Mv2, consisting of 90M+ nodes and 600M+ edges. Our solution uses a diverse ensemble of $85$ Knowledge Graph Embedding models combining five different scoring functions (TransE, TransH, RotatE, DistMult, ComplEx) and two different loss functions (log-sigmoid, sampled softmax cross-entropy). Each individual model is trained in parallel on a Graphcore Bow Pod$_{16}$ using BESS (Balanced Entity Sampling and Sharing), a new distribution framework for KGE training and inference based on balanced collective communications between workers. Our final model achieves a validation MRR of 0.2922 and a test-challenge MRR of 0.2562, winning the first place in the competition. The code is publicly available at: https://github.com/graphcore/distributed-kge-poplar/tree/2022-ogb-submission.