 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOntoMerger: An Ontology Integration Library for Deduplicating and Connecting Knowledge Graph Nodes
Jun 05, 2022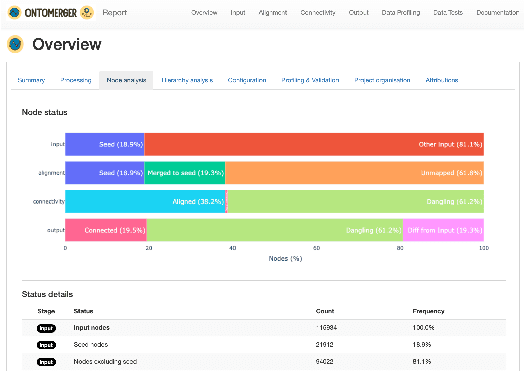

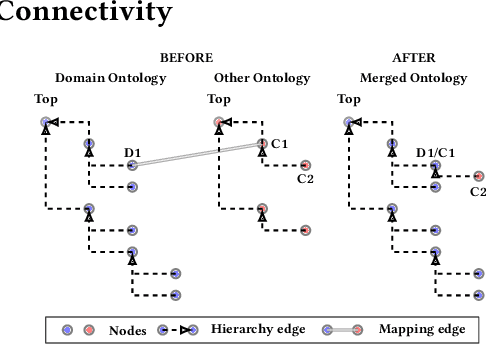

Duplication of nodes is a common problem encountered when building knowledge graphs (KGs) from heterogeneous datasets, where it is crucial to be able to merge nodes having the same meaning. OntoMerger is a Python ontology integration library whose functionality is to deduplicate KG nodes. Our approach takes a set of KG nodes, mappings and disconnected hierarchies and generates a set of merged nodes together with a connected hierarchy. In addition, the library provides analytic and data testing functionalities that can be used to fine-tune the inputs, further reducing duplication, and to increase connectivity of the output graph. OntoMerger can be applied to a wide variety of ontologies and KGs. In this paper we introduce OntoMerger and illustrate its functionality on a real-world biomedical KG.
ChemicalX: A Deep Learning Library for Drug Pair Scoring
Feb 14, 2022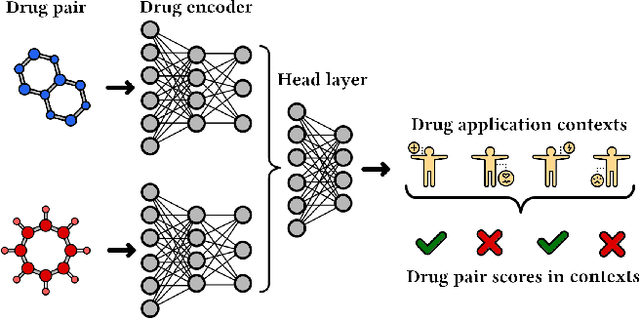
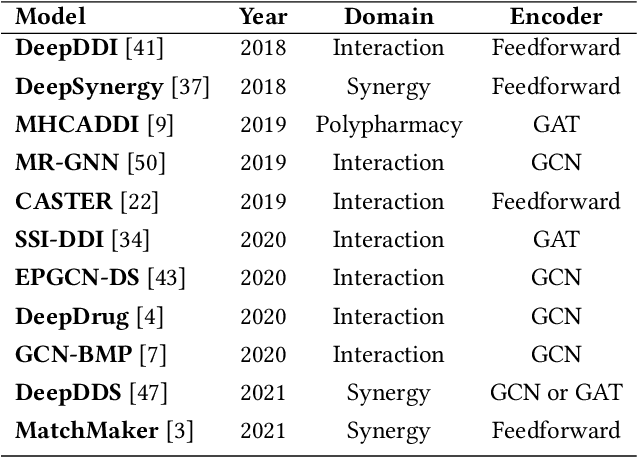
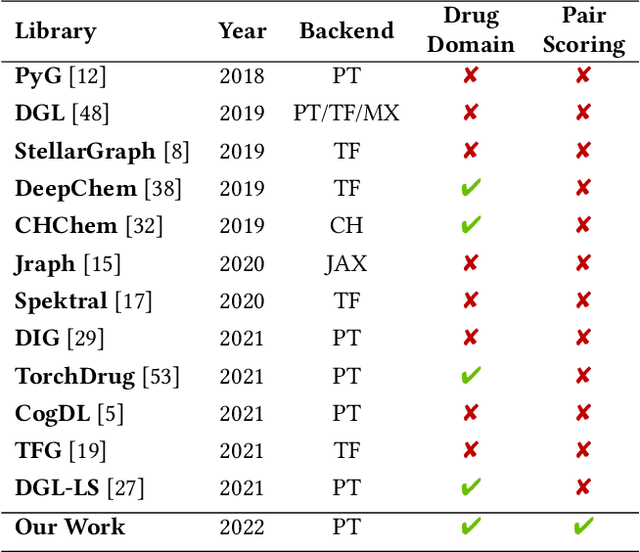
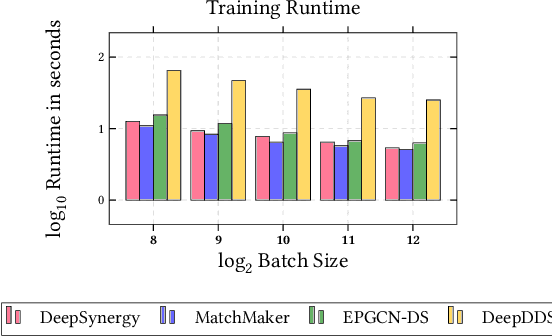
In this paper, we introduce ChemicalX, a PyTorch-based deep learning library designed for providing a range of state of the art models to solve the drug pair scoring task. The primary objective of the library is to make deep drug pair scoring models accessible to machine learning researchers and practitioners in a streamlined framework.The design of ChemicalX reuses existing high level model training utilities, geometric deep learning, and deep chemistry layers from the PyTorch ecosystem. Our system provides neural network layers, custom pair scoring architectures, data loaders, and batch iterators for end users. We showcase these features with example code snippets and case studies to highlight the characteristics of ChemicalX. A range of experiments on real world drug-drug interaction, polypharmacy side effect, and combination synergy prediction tasks demonstrate that the models available in ChemicalX are effective at solving the pair scoring task. Finally, we show that ChemicalX could be used to train and score machine learning models on large drug pair datasets with hundreds of thousands of compounds on commodity hardware.
A Unified View of Relational Deep Learning for Drug Pair Scoring
Nov 14, 2021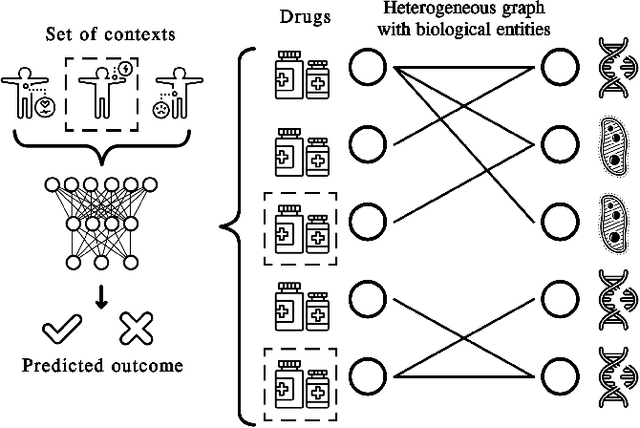
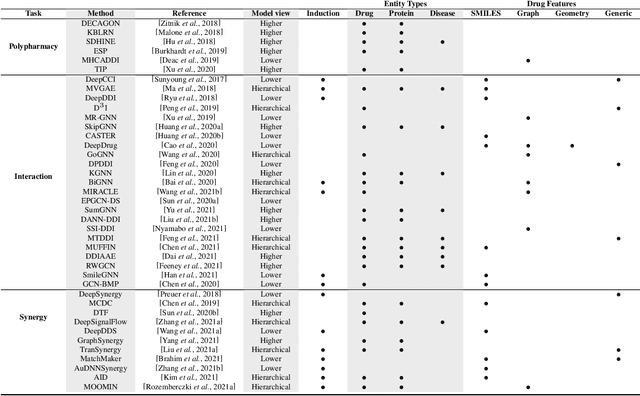
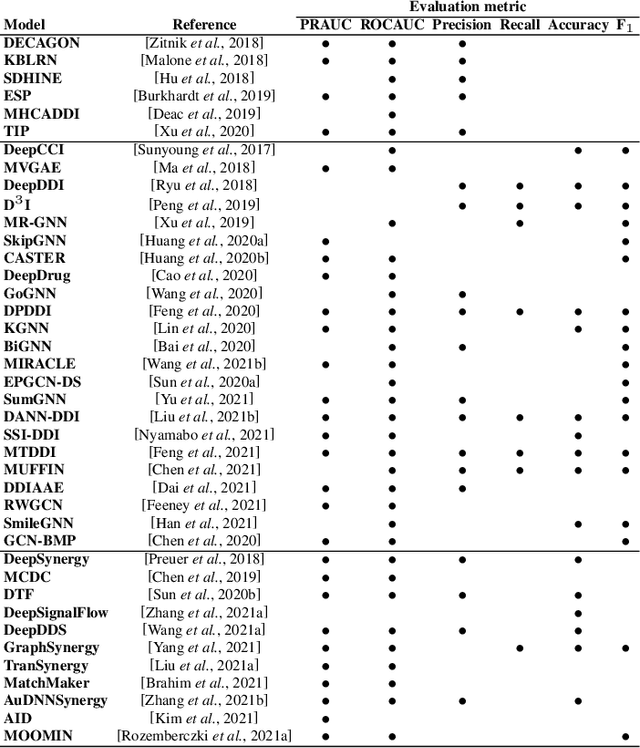
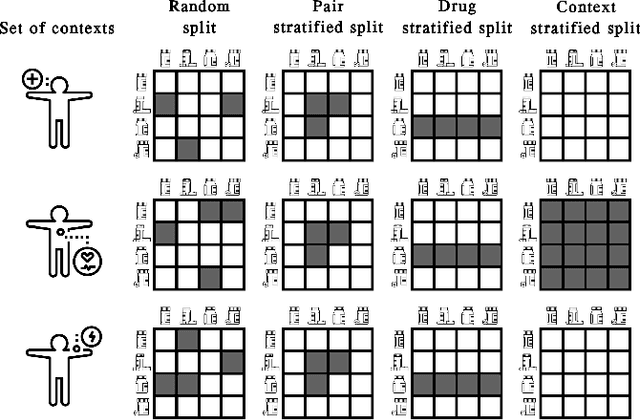
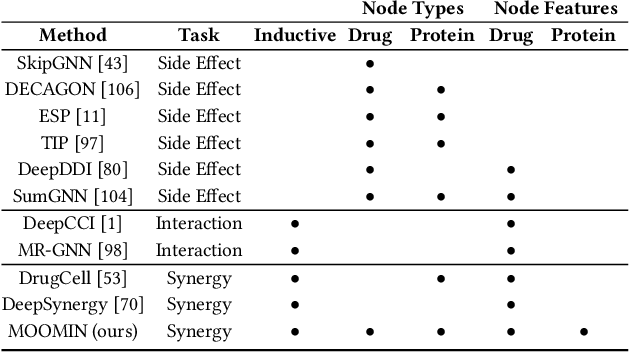
In recent years, numerous machine learning models which attempt to solve polypharmacy side effect identification, drug-drug interaction prediction and combination therapy design tasks have been proposed. Here, we present a unified theoretical view of relational machine learning models which can address these tasks. We provide fundamental definitions, compare existing model architectures and discuss performance metrics, datasets and evaluation protocols. In addition, we emphasize possible high impact applications and important future research directions in this domain.
MOOMIN: Deep Molecular Omics Network for Anti-Cancer Drug Combination Therapy
Oct 28, 2021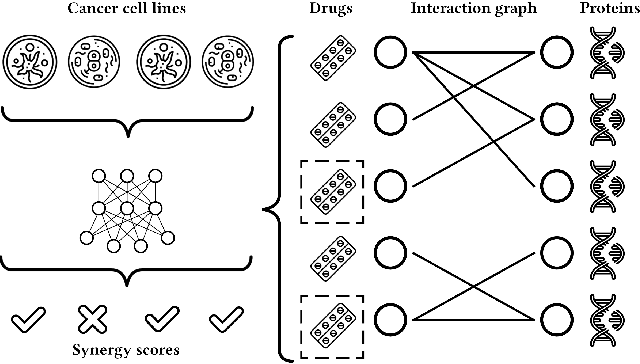
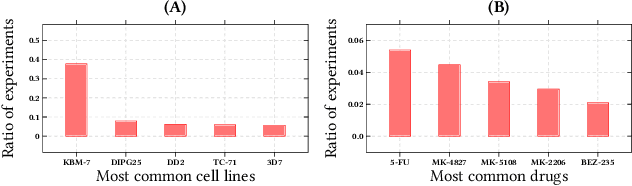

We propose the molecular omics network (MOOMIN) a multimodal graph neural network that can predict the synergistic effect of drug combinations for cancer treatment. Our model captures the representation based on the context of drugs at multiple scales based on a drug-protein interaction network and metadata. Structural properties of the compounds and proteins are encoded to create vertex features for a message-passing scheme that operates on the bipartite interaction graph. Propagated messages form multi-resolution drug representations which we utilized to create drug pair descriptors. By conditioning the drug combination representations on the cancer cell type we define a synergy scoring function that can inductively score unseen pairs of drugs. Experimental results on the synergy scoring task demonstrate that MOOMIN outperforms state-of-the-art graph fingerprinting, proximity preserving node embedding, and existing deep learning approaches. Further results establish that the predictive performance of our model is robust to hyperparameter changes. We demonstrate that the model makes high-quality predictions over a wide range of cancer cell line tissues, out-of-sample predictions can be validated with external synergy databases, and that the proposed model is data-efficient at learning.