 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Neural Networks for Microbial Genome Recovery
Apr 26, 2022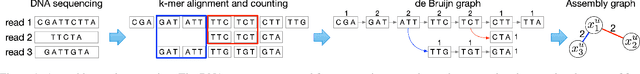
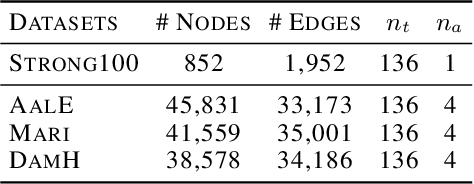
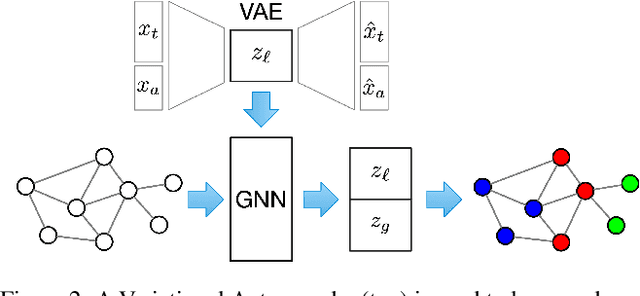
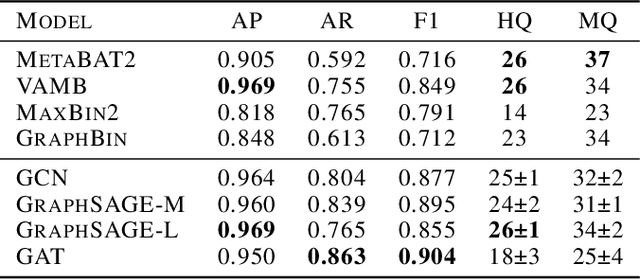
Microbes have a profound impact on our health and environment, but our understanding of the diversity and function of microbial communities is severely limited. Through DNA sequencing of microbial communities (metagenomics), DNA fragments (reads) of the individual microbes can be obtained, which through assembly graphs can be combined into long contiguous DNA sequences (contigs). Given the complexity of microbial communities, single contig microbial genomes are rarely obtained. Instead, contigs are eventually clustered into bins, with each bin ideally making up a full genome. This process is referred to as metagenomic binning. Current state-of-the-art techniques for metagenomic binning rely only on the local features for the individual contigs. These techniques therefore fail to exploit the similarities between contigs as encoded by the assembly graph, in which the contigs are organized. In this paper, we propose to use Graph Neural Networks (GNNs) to leverage the assembly graph when learning contig representations for metagenomic binning. Our method, VaeG-Bin, combines variational autoencoders for learning latent representations of the individual contigs, with GNNs for refining these representations by taking into account the neighborhood structure of the contigs in the assembly graph. We explore several types of GNNs and demonstrate that VaeG-Bin recovers more high-quality genomes than other state-of-the-art binners on both simulated and real-world datasets.
Priberam Labs at the NTCIR-15 SHINRA2020-ML: Classification Task
May 12, 2021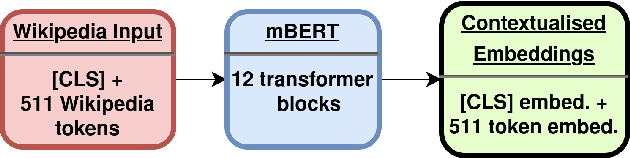
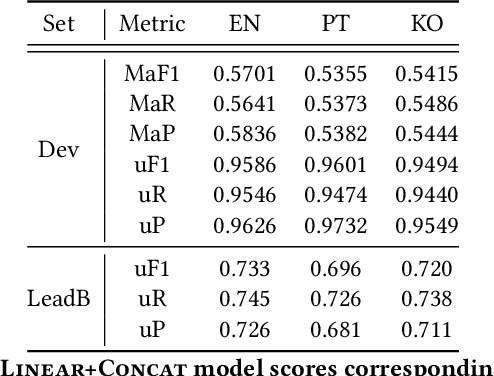
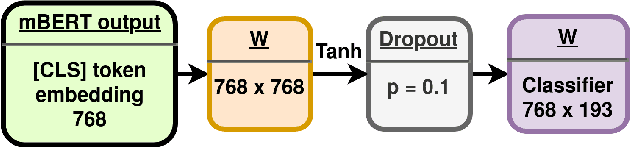
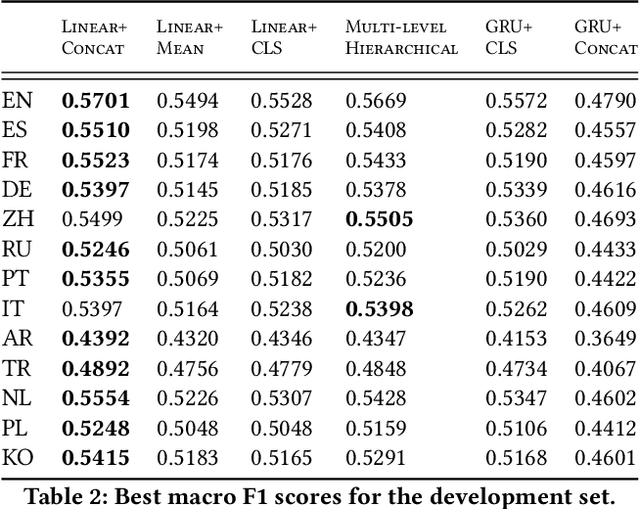
Wikipedia is an online encyclopedia available in 285 languages. It composes an extremely relevant Knowledge Base (KB), which could be leveraged by automatic systems for several purposes. However, the structure and organisation of such information are not prone to automatic parsing and understanding and it is, therefore, necessary to structure this knowledge. The goal of the current SHINRA2020-ML task is to leverage Wikipedia pages in order to categorise their corresponding entities across 268 hierarchical categories, belonging to the Extended Named Entity (ENE) ontology. In this work, we propose three distinct models based on the contextualised embeddings yielded by Multilingual BERT. We explore the performances of a linear layer with and without explicit usage of the ontology's hierarchy, and a Gated Recurrent Units (GRU) layer. We also test several pooling strategies to leverage BERT's embeddings and selection criteria based on the labels' scores. We were able to achieve good performance across a large variety of languages, including those not seen during the fine-tuning process (zero-shot languages).
Using Neural Networks for Relation Extraction from Biomedical Literature
May 27, 2019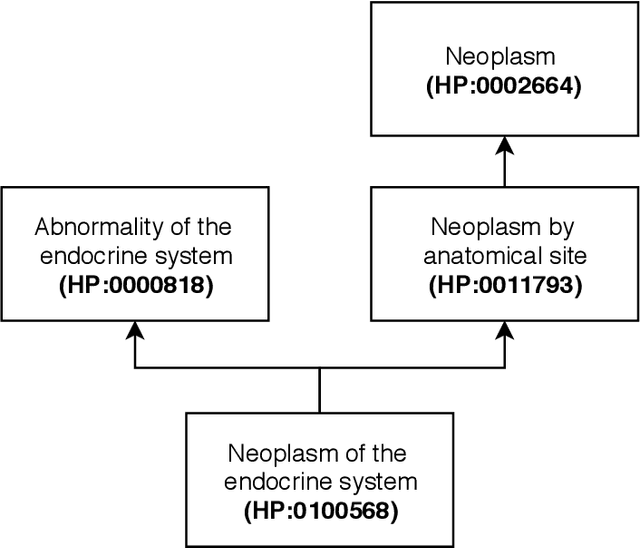

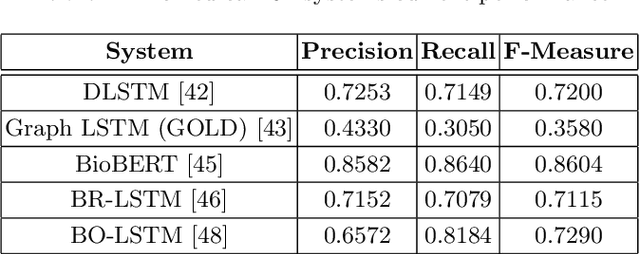
Using different sources of information to support automated extracting of relations between biomedical concepts contributes to the development of our understanding of biological systems. The primary comprehensive source of these relations is biomedical literature. Several relation extraction approaches have been proposed to identify relations between concepts in biomedical literature, namely using neural networks algorithms. The use of multichannel architectures composed of multiple data representations, as in deep neural networks, is leading to state-of-the-art results. The right combination of data representations can eventually lead us to even higher evaluation scores in relation extraction tasks. Thus, biomedical ontologies play a fundamental role by providing semantic and ancestry information about an entity. The incorporation of biomedical ontologies has already been proved to enhance previous state-of-the-art results.
A Silver Standard Corpus of Human Phenotype-Gene Relations
Mar 26, 2019


Human phenotype-gene relations are fundamental to fully understand the origin of some phenotypic abnormalities and their associated diseases. Biomedical literature is the most comprehensive source of these relations, however, we need Relation Extraction tools to automatically recognize them. Most of these tools require an annotated corpus and to the best of our knowledge, there is no corpus available annotated with human phenotype-gene relations. This paper presents the Phenotype-Gene Relations (PGR) corpus, a silver standard corpus of human phenotype and gene annotations and their relations. The corpus consists of 1712 abstracts, 5676 human phenotype annotations, 13835 gene annotations, and 4283 relations. We generated this corpus using Named-Entity Recognition tools, whose results were partially evaluated by eight curators, obtaining a precision of 87.01%. By using the corpus we were able to obtain promising results with two state-of-the-art deep learning tools, namely 78.05% of precision. The PGR corpus was made publicly available to the research community.
WS4A: a Biomedical Question and Answering System based on public Web Services and Ontologies
Nov 17, 2016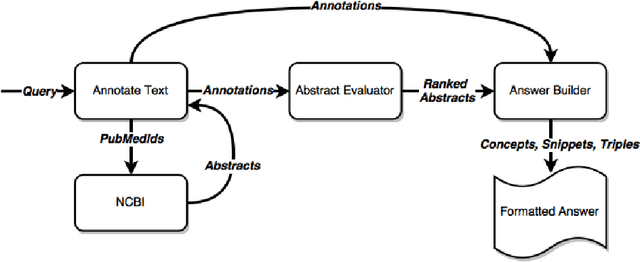
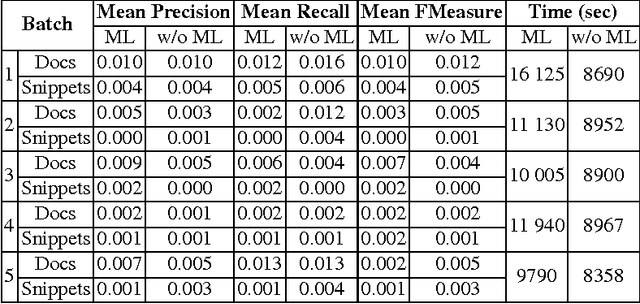
This paper describes our system, dubbed WS4A (Web Services for All), that participated in the fourth edition of the BioASQ challenge (2016). We used WS4A to perform the Question and Answering (QA) task 4b, which consisted on the retrieval of relevant concepts, documents, snippets, RDF triples, exact answers and ideal answers for each given question. The novelty in our approach consists on the maximum exploitation of existing web services in each step of WS4A, such as the annotation of text, and the retrieval of metadata for each annotation. The information retrieved included concept identifiers, ontologies, ancestors, and most importantly, PubMed identifiers. The paper describes the WS4A pipeline and also presents the precision, recall and f-measure values obtained in task 4b. Our system achieved two second places in two subtasks on one of the five batches.