 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFASTR: Reimagining FASTQ via Compact Image-inspired Representation
Jan 23, 2026Motivation: High-throughput sequencing (HTS) enables population-scale genomics but generates massive datasets, creating bottlenecks in storage, transfer, and analysis. FASTQ, the standard format for over two decades, stores one byte per base and one byte per quality score, leading to inefficient I/O, high storage costs, and redundancy. Existing compression tools can mitigate some issues, but often introduce costly decompression or complex dependency issues. Results: We introduce FASTR, a lossless, computation-native successor to FASTQ that encodes each nucleotide together with its base quality score into a single 8-bit value. FASTR reduces file size by at least 2x while remaining fully reversible and directly usable for downstream analyses. Applying general-purpose compression tools on FASTR consistently yields higher compression ratios, 2.47, 3.64, and 4.8x faster compression, and 2.34, 1.96, 1.75x faster decompression than on FASTQ across Illumina, HiFi, and ONT reads. FASTR is machine-learning-ready, allowing reads to be consumed directly as numerical vectors or image-like representations. We provide a highly parallel software ecosystem for FASTQ-FASTR conversion and show that FASTR integrates with existing tools, such as minimap2, with minimal interface changes and no performance overhead. By eliminating decompression costs and reducing data movement, FASTR lays the foundation for scalable genomics analyses and real-time sequencing workflows. Availability and Implementation: https://github.com/ALSER-Lab/FASTR
Efficient Approximate Kernel Based Spike Sequence Classification
Sep 11, 2022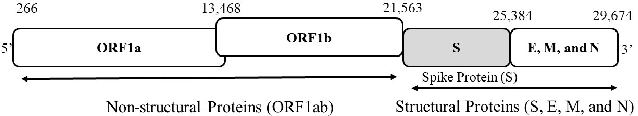
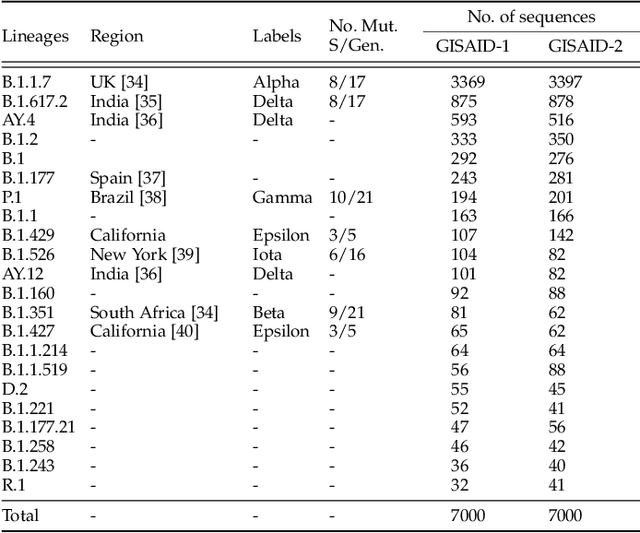
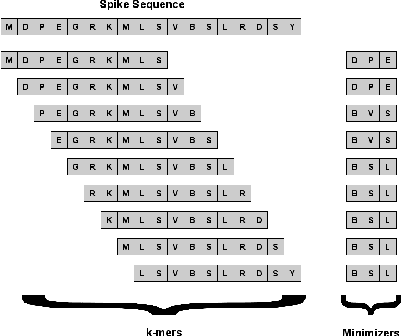
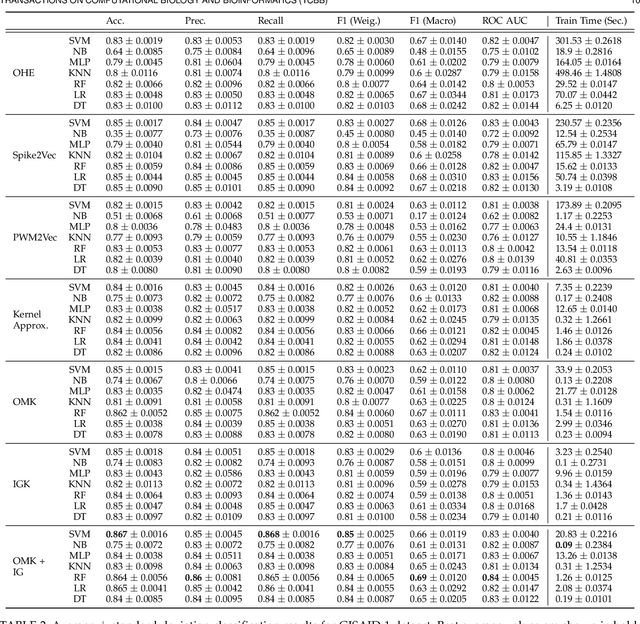
Machine learning (ML) models, such as SVM, for tasks like classification and clustering of sequences, require a definition of distance/similarity between pairs of sequences. Several methods have been proposed to compute the similarity between sequences, such as the exact approach that counts the number of matches between $k$-mers (sub-sequences of length $k$) and an approximate approach that estimates pairwise similarity scores. Although exact methods yield better classification performance, they pose high computational costs, limiting their applicability to a small number of sequences. The approximate algorithms are proven to be more scalable and perform comparably to (sometimes better than) the exact methods -- they are designed in a "general" way to deal with different types of sequences (e.g., music, protein, etc.). Although general applicability is a desired property of an algorithm, it is not the case in all scenarios. For example, in the current COVID-19 (coronavirus) pandemic, there is a need for an approach that can deal specifically with the coronavirus. To this end, we propose a series of ways to improve the performance of the approximate kernel (using minimizers and information gain) in order to enhance its predictive performance pm coronavirus sequences. More specifically, we improve the quality of the approximate kernel using domain knowledge (computed using information gain) and efficient preprocessing (using minimizers computation) to classify coronavirus spike protein sequences corresponding to different variants (e.g., Alpha, Beta, Gamma). We report results using different classification and clustering algorithms and evaluate their performance using multiple evaluation metrics. Using two datasets, we show that our proposed method helps improve the kernel's performance compared to the baseline and state-of-the-art approaches in the healthcare domain.