 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLIMBA: An Open-Source Framework for the Preservation and Valorization of Low-Resource Languages using Generative Models
Nov 20, 2024

Minority languages are vital to preserving cultural heritage, yet they face growing risks of extinction due to limited digital resources and the dominance of artificial intelligence models trained on high-resource languages. This white paper proposes a framework to generate linguistic tools for low-resource languages, focusing on data creation to support the development of language models that can aid in preservation efforts. Sardinian, an endangered language, serves as the case study to demonstrate the framework's effectiveness. By addressing the data scarcity that hinders intelligent applications for such languages, we contribute to promoting linguistic diversity and support ongoing efforts in language standardization and revitalization through modern technologies.
Iterative Zero-Shot LLM Prompting for Knowledge Graph Construction
Jul 03, 2023In the current digitalization era, capturing and effectively representing knowledge is crucial in most real-world scenarios. In this context, knowledge graphs represent a potent tool for retrieving and organizing a vast amount of information in a properly interconnected and interpretable structure. However, their generation is still challenging and often requires considerable human effort and domain expertise, hampering the scalability and flexibility across different application fields. This paper proposes an innovative knowledge graph generation approach that leverages the potential of the latest generative large language models, such as GPT-3.5, that can address all the main critical issues in knowledge graph building. The approach is conveyed in a pipeline that comprises novel iterative zero-shot and external knowledge-agnostic strategies in the main stages of the generation process. Our unique manifold approach may encompass significant benefits to the scientific community. In particular, the main contribution can be summarized by: (i) an innovative strategy for iteratively prompting large language models to extract relevant components of the final graph; (ii) a zero-shot strategy for each prompt, meaning that there is no need for providing examples for "guiding" the prompt result; (iii) a scalable solution, as the adoption of LLMs avoids the need for any external resources or human expertise. To assess the effectiveness of our proposed model, we performed experiments on a dataset that covered a specific domain. We claim that our proposal is a suitable solution for scalable and versatile knowledge graph construction and may be applied to different and novel contexts.
Characterization of graphs for protein structure modeling and recognition of solubility
Sep 23, 2015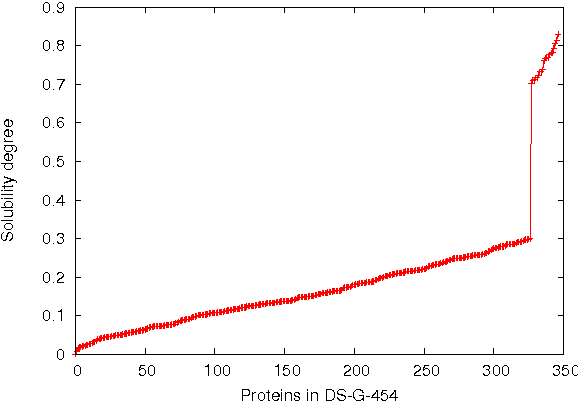
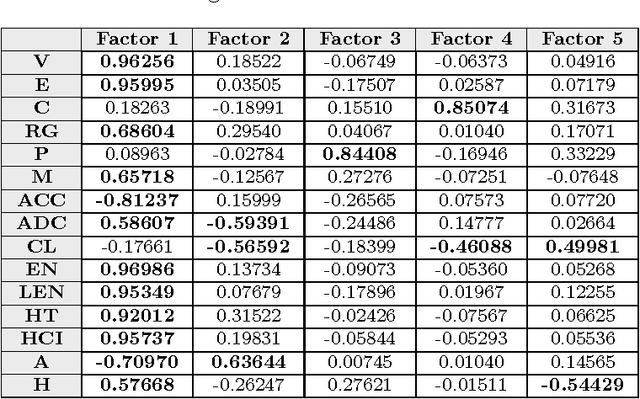
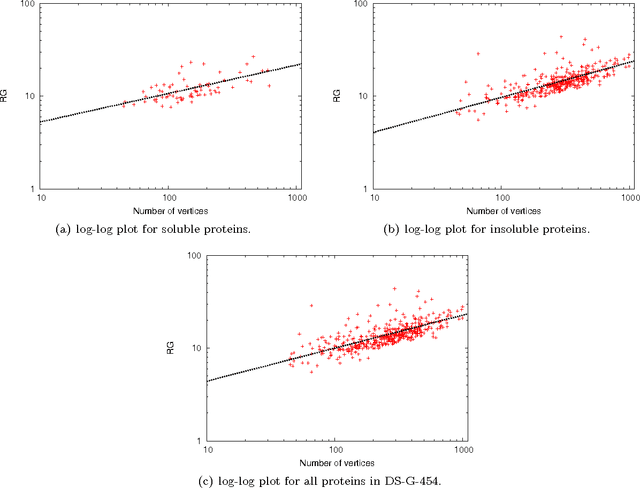
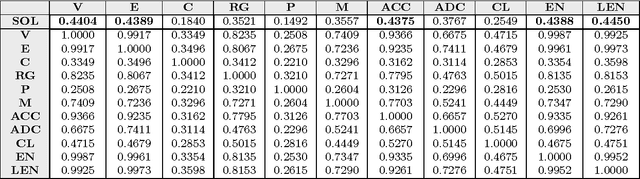
This paper deals with the relations among structural, topological, and chemical properties of the E.Coli proteome from the vantage point of the solubility/aggregation propensity of proteins. Each E.Coli protein is initially represented according to its known folded 3D shape. This step consists in representing the available E.Coli proteins in terms of graphs. We first analyze those graphs by considering pure topological characterizations, i.e., by analyzing the mass fractal dimension and the distribution underlying both shortest paths and vertex degrees. Results confirm the general architectural principles of proteins. Successively, we focus on the statistical properties of a representation of such graphs in terms of vectors composed of several numerical features, which we extracted from their structural representation. We found that protein size is the main discriminator for the solubility, while however there are other factors that help explaining the solubility degree. We finally analyze such data through a novel one-class classifier, with the aim of discriminating among very and poorly soluble proteins. Results are encouraging and consolidate the potential of pattern recognition techniques when employed to describe complex biological systems.
Toward a multilevel representation of protein molecules: comparative approaches to the aggregation/folding propensity problem
Apr 30, 2015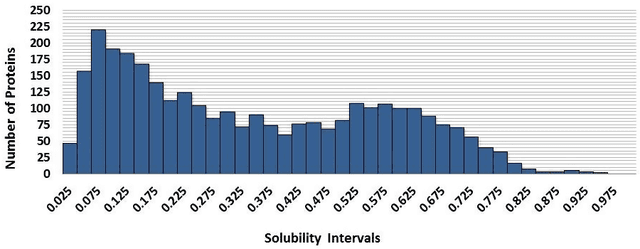
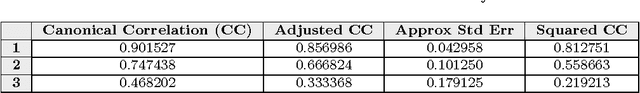
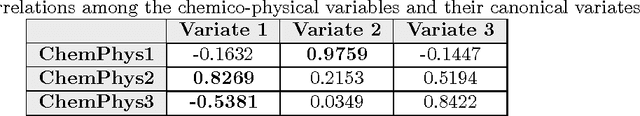
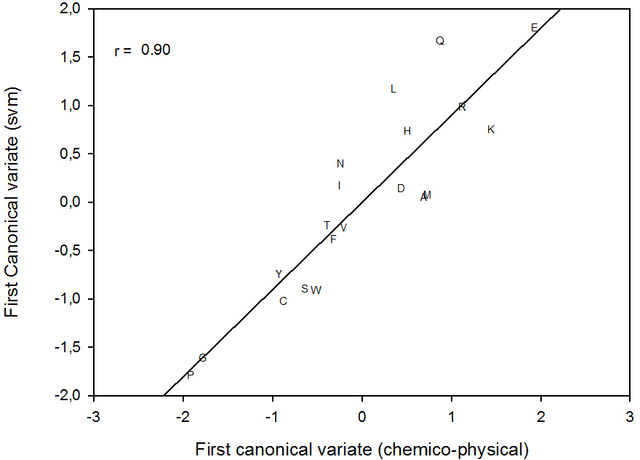
This paper builds upon the fundamental work of Niwa et al. [34], which provides the unique possibility to analyze the relative aggregation/folding propensity of the elements of the entire Escherichia coli (E. coli) proteome in a cell-free standardized microenvironment. The hardness of the problem comes from the superposition between the driving forces of intra- and inter-molecule interactions and it is mirrored by the evidences of shift from folding to aggregation phenotypes by single-point mutations [10]. Here we apply several state-of-the-art classification methods coming from the field of structural pattern recognition, with the aim to compare different representations of the same proteins gathered from the Niwa et al. data base; such representations include sequences and labeled (contact) graphs enriched with chemico-physical attributes. By this comparison, we are able to identify also some interesting general properties of proteins. Notably, (i) we suggest a threshold around 250 residues discriminating "easily foldable" from "hardly foldable" molecules consistent with other independent experiments, and (ii) we highlight the relevance of contact graph spectra for folding behavior discrimination and characterization of the E. coli solubility data. The soundness of the experimental results presented in this paper is proved by the statistically relevant relationships discovered among the chemico-physical description of proteins and the developed cost matrix of substitution used in the various discrimination systems.