 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeToward a multilevel representation of protein molecules: comparative approaches to the aggregation/folding propensity problem
Paper and Code
Apr 30, 2015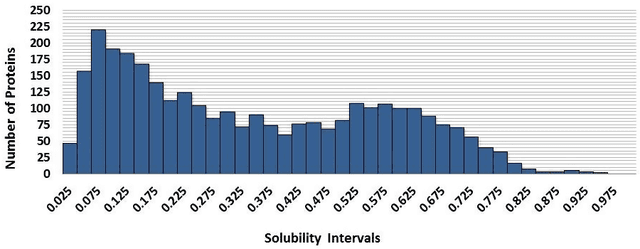
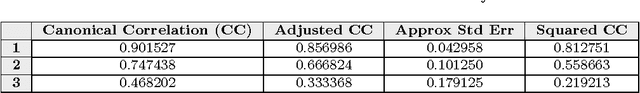
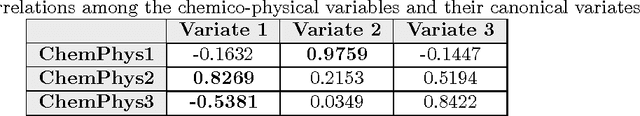
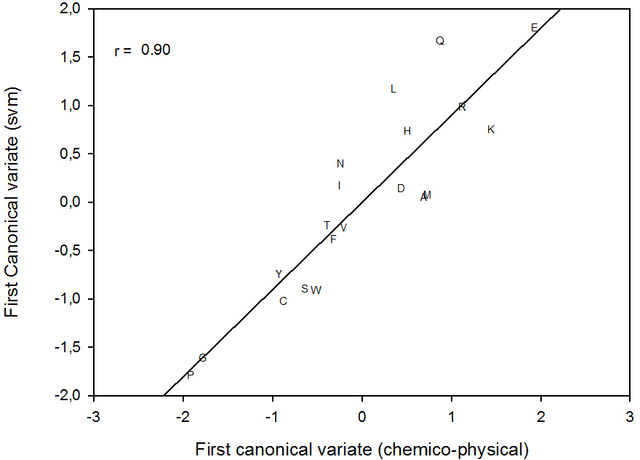
This paper builds upon the fundamental work of Niwa et al. [34], which provides the unique possibility to analyze the relative aggregation/folding propensity of the elements of the entire Escherichia coli (E. coli) proteome in a cell-free standardized microenvironment. The hardness of the problem comes from the superposition between the driving forces of intra- and inter-molecule interactions and it is mirrored by the evidences of shift from folding to aggregation phenotypes by single-point mutations [10]. Here we apply several state-of-the-art classification methods coming from the field of structural pattern recognition, with the aim to compare different representations of the same proteins gathered from the Niwa et al. data base; such representations include sequences and labeled (contact) graphs enriched with chemico-physical attributes. By this comparison, we are able to identify also some interesting general properties of proteins. Notably, (i) we suggest a threshold around 250 residues discriminating "easily foldable" from "hardly foldable" molecules consistent with other independent experiments, and (ii) we highlight the relevance of contact graph spectra for folding behavior discrimination and characterization of the E. coli solubility data. The soundness of the experimental results presented in this paper is proved by the statistically relevant relationships discovered among the chemico-physical description of proteins and the developed cost matrix of substitution used in the various discrimination systems.