 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTEPI: Taxonomy-aware Embedding and Pseudo-Imaging for Scarcely-labeled Zero-shot Genome Classification
Jan 24, 2024A species' genetic code or genome encodes valuable evolutionary, biological, and phylogenetic information that aids in species recognition, taxonomic classification, and understanding genetic predispositions like drug resistance and virulence. However, the vast number of potential species poses significant challenges in developing a general-purpose whole genome classification tool. Traditional bioinformatics tools have made notable progress but lack scalability and are computationally expensive. Machine learning-based frameworks show promise but must address the issue of large classification vocabularies with long-tail distributions. In this study, we propose addressing this problem through zero-shot learning using TEPI, Taxonomy-aware Embedding and Pseudo-Imaging. We represent each genome as pseudo-images and map them to a taxonomy-aware embedding space for reasoning and classification. This embedding space captures compositional and phylogenetic relationships of species, enabling predictions in extensive search spaces. We evaluate TEPI using two rigorous zero-shot settings and demonstrate its generalization capabilities qualitatively on curated, large-scale, publicly sourced data.
Scalable Pathogen Detection from Next Generation DNA Sequencing with Deep Learning
Nov 30, 2022Next-generation sequencing technologies have enhanced the scope of Internet-of-Things (IoT) to include genomics for personalized medicine through the increased availability of an abundance of genome data collected from heterogeneous sources at a reduced cost. Given the sheer magnitude of the collected data and the significant challenges offered by the presence of highly similar genomic structure across species, there is a need for robust, scalable analysis platforms to extract actionable knowledge such as the presence of potentially zoonotic pathogens. The emergence of zoonotic diseases from novel pathogens, such as the influenza virus in 1918 and SARS-CoV-2 in 2019 that can jump species barriers and lead to pandemic underscores the need for scalable metagenome analysis. In this work, we propose MG2Vec, a deep learning-based solution that uses the transformer network as its backbone, to learn robust features from raw metagenome sequences for downstream biomedical tasks such as targeted and generalized pathogen detection. Extensive experiments on four increasingly challenging, yet realistic diagnostic settings, show that the proposed approach can help detect pathogens from uncurated, real-world clinical samples with minimal human supervision in the form of labels. Further, we demonstrate that the learned representations can generalize to completely unrelated pathogens across diseases and species for large-scale metagenome analysis. We provide a comprehensive evaluation of a novel representation learning framework for metagenome-based disease diagnostics with deep learning and provide a way forward for extracting and using robust vector representations from low-cost next generation sequencing to develop generalizable diagnostic tools.
Metagenome2Vec: Building Contextualized Representations for Scalable Metagenome Analysis
Nov 09, 2021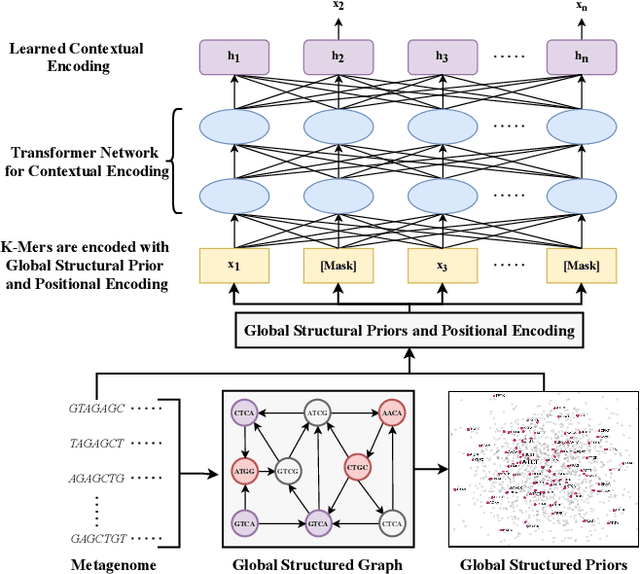
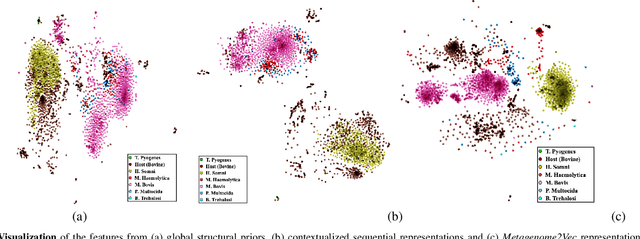
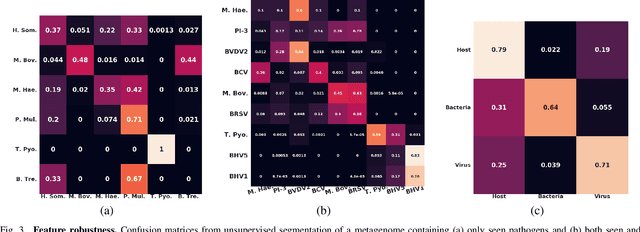
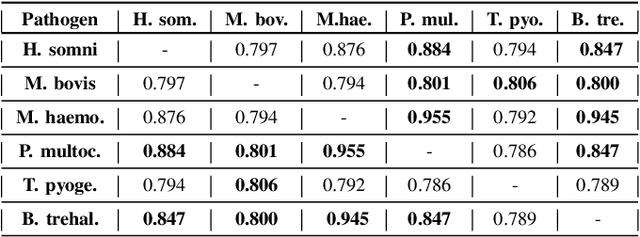
Advances in next-generation metagenome sequencing have the potential to revolutionize the point-of-care diagnosis of novel pathogen infections, which could help prevent potential widespread transmission of diseases. Given the high volume of metagenome sequences, there is a need for scalable frameworks to analyze and segment metagenome sequences from clinical samples, which can be highly imbalanced. There is an increased need for learning robust representations from metagenome reads since pathogens within a family can have highly similar genome structures (some more than 90%) and hence enable the segmentation and identification of novel pathogen sequences with limited labeled data. In this work, we propose Metagenome2Vec - a contextualized representation that captures the global structural properties inherent in metagenome data and local contextualized properties through self-supervised representation learning. We show that the learned representations can help detect six (6) related pathogens from clinical samples with less than 100 labeled sequences. Extensive experiments on simulated and clinical metagenome data show that the proposed representation encodes compositional properties that can generalize beyond annotations to segment novel pathogens in an unsupervised setting.
MG-NET: Leveraging Pseudo-Imaging for Multi-Modal Metagenome Analysis
Jul 21, 2021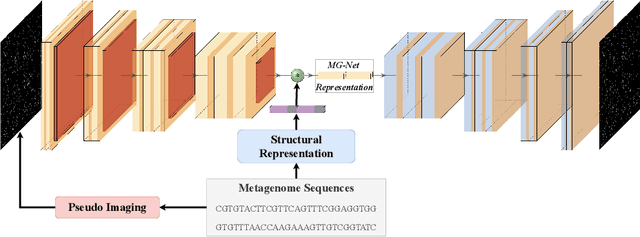
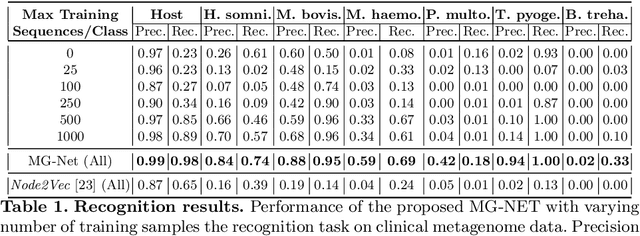
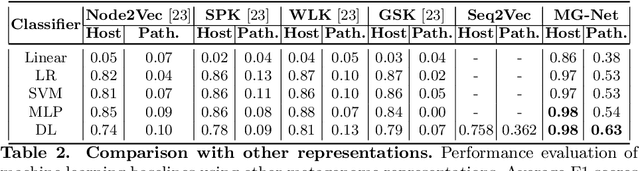
The emergence of novel pathogens and zoonotic diseases like the SARS-CoV-2 have underlined the need for developing novel diagnosis and intervention pipelines that can learn rapidly from small amounts of labeled data. Combined with technological advances in next-generation sequencing, metagenome-based diagnostic tools hold much promise to revolutionize rapid point-of-care diagnosis. However, there are significant challenges in developing such an approach, the chief among which is to learn self-supervised representations that can help detect novel pathogen signatures with very low amounts of labeled data. This is particularly a difficult task given that closely related pathogens can share more than 90% of their genome structure. In this work, we address these challenges by proposing MG-Net, a self-supervised representation learning framework that leverages multi-modal context using pseudo-imaging data derived from clinical metagenome sequences. We show that the proposed framework can learn robust representations from unlabeled data that can be used for downstream tasks such as metagenome sequence classification with limited access to labeled data. Extensive experiments show that the learned features outperform current baseline metagenome representations, given only 1000 samples per class.
Genome Sequence Classification for Animal Diagnostics with Graph Representations and Deep Neural Networks
Jul 24, 2020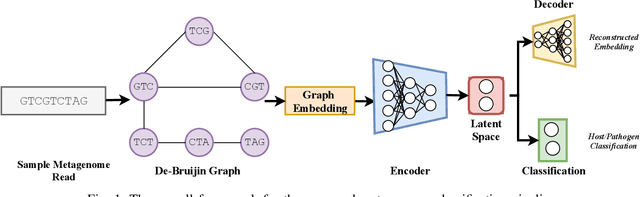
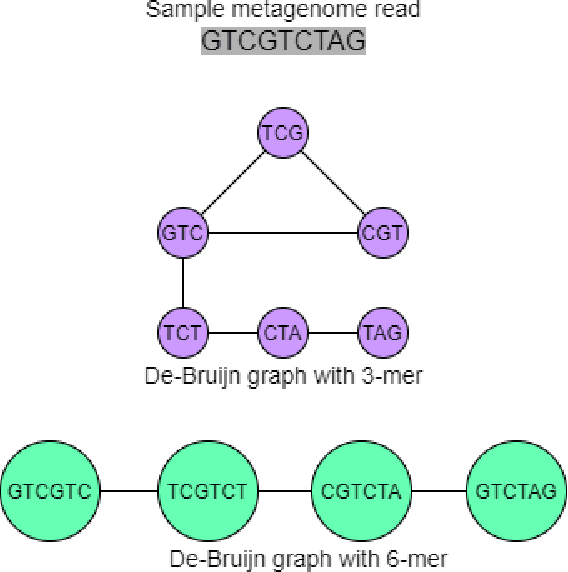
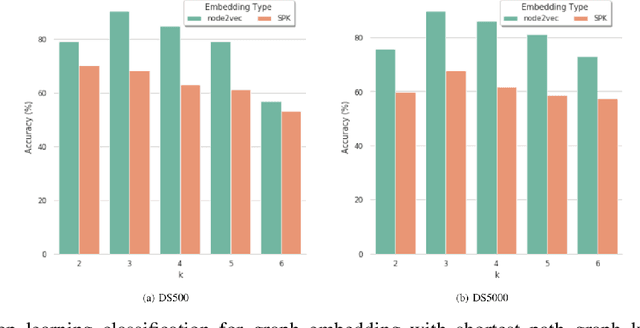
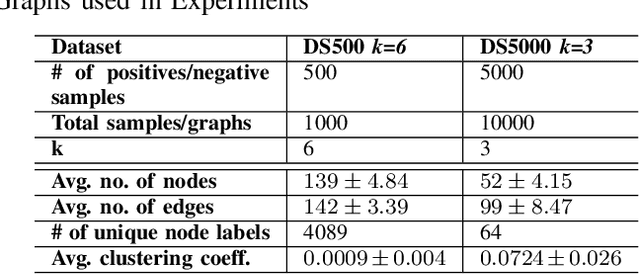
Bovine Respiratory Disease Complex (BRDC) is a complex respiratory disease in cattle with multiple etiologies, including bacterial and viral. It is estimated that mortality, morbidity, therapy, and quarantine resulting from BRDC account for significant losses in the cattle industry. Early detection and management of BRDC are crucial in mitigating economic losses. Current animal disease diagnostics is based on traditional tests such as bacterial culture, serolog, and Polymerase Chain Reaction (PCR) tests. Even though these tests are validated for several diseases, their main challenge is their limited ability to detect the presence of multiple pathogens simultaneously. Advancements of data analytics and machine learning and applications over metagenome sequencing are setting trends on several applications. In this work, we demonstrate a machine learning approach to identify pathogen signatures present in bovine metagenome sequences using k-mer-based network embedding followed by a deep learning-based classification task. With experiments conducted on two different simulated datasets, we show that networks-based machine learning approaches can detect pathogen signature with up to 89.7% accuracy. We will make the data available publicly upon request to tackle this important problem in a difficult domain.