 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIndependent SE-Equivariant Models for End-to-End Rigid Protein Docking
Paper and Code
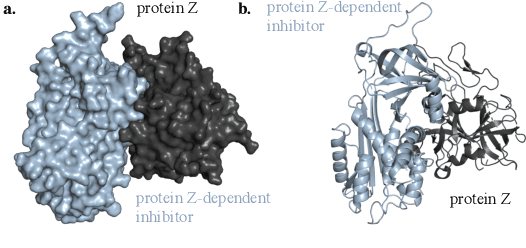
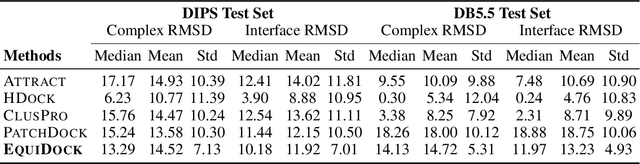
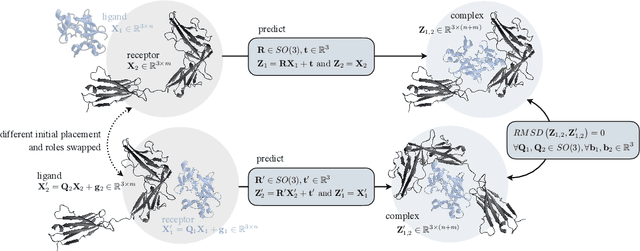

Protein complex formation is a central problem in biology, being involved in most of the cell's processes, and essential for applications, e.g. drug design or protein engineering. We tackle rigid body protein-protein docking, i.e., computationally predicting the 3D structure of a protein-protein complex from the individual unbound structures, assuming no conformational change within the proteins happens during binding. We design a novel pairwise-independent SE(3)-equivariant graph matching network to predict the rotation and translation to place one of the proteins at the right docked position relative to the second protein. We mathematically guarantee a basic principle: the predicted complex is always identical regardless of the initial locations and orientations of the two structures. Our model, named EquiDock, approximates the binding pockets and predicts the docking poses using keypoint matching and alignment, achieved through optimal transport and a differentiable Kabsch algorithm. Empirically, we achieve significant running time improvements and often outperform existing docking software despite not relying on heavy candidate sampling, structure refinement, or templates.