 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge3D Deep Learning for Biological Function Prediction from Physical Fields
Paper and Code
Apr 13, 2017
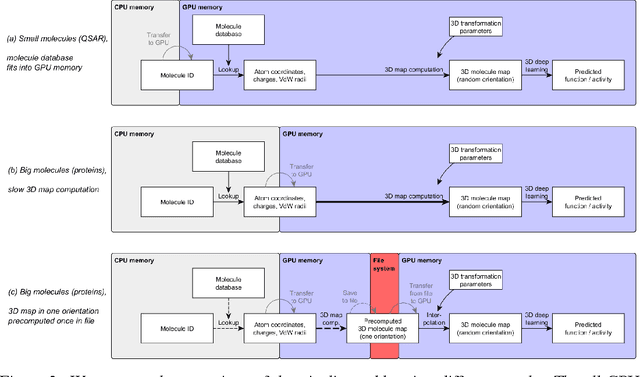
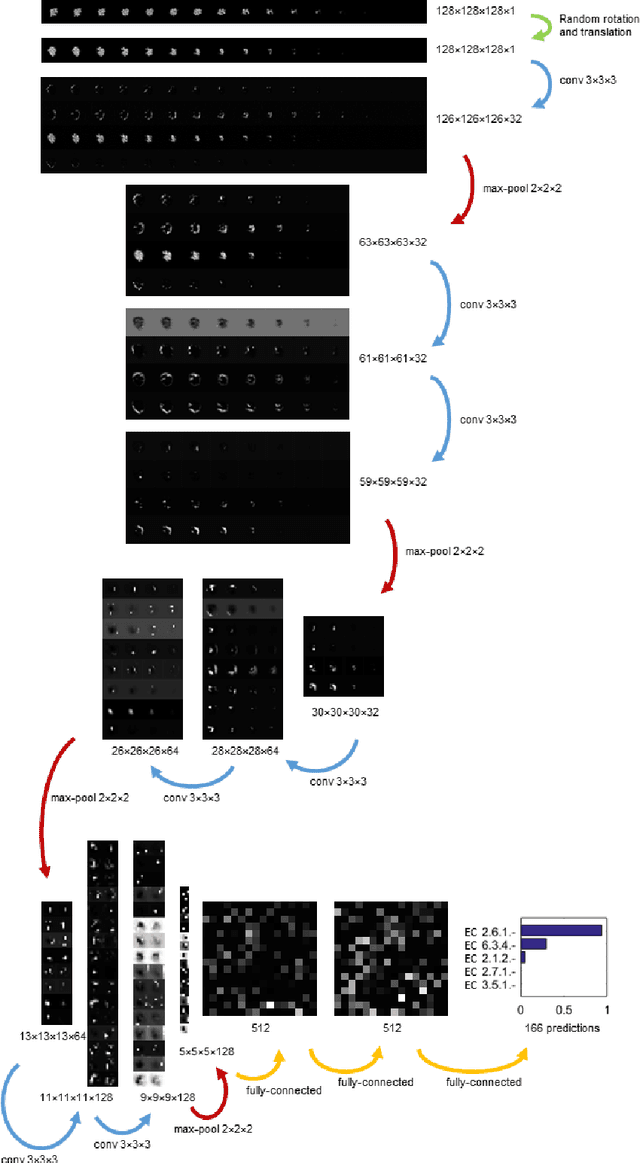
Predicting the biological function of molecules, be it proteins or drug-like compounds, from their atomic structure is an important and long-standing problem. Function is dictated by structure, since it is by spatial interactions that molecules interact with each other, both in terms of steric complementarity, as well as intermolecular forces. Thus, the electron density field and electrostatic potential field of a molecule contain the "raw fingerprint" of how this molecule can fit to binding partners. In this paper, we show that deep learning can predict biological function of molecules directly from their raw 3D approximated electron density and electrostatic potential fields. Protein function based on EC numbers is predicted from the approximated electron density field. In another experiment, the activity of small molecules is predicted with quality comparable to state-of-the-art descriptor-based methods. We propose several alternative computational models for the GPU with different memory and runtime requirements for different sizes of molecules and of databases. We also propose application-specific multi-channel data representations. With future improvements of training datasets and neural network settings in combination with complementary information sources (sequence, genomic context, expression level), deep learning can be expected to show its generalization power and revolutionize the field of molecular function prediction.