 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAnalysis of ensemble feature selection for correlated high-dimensional RNA-Seq cancer data
Apr 28, 2020

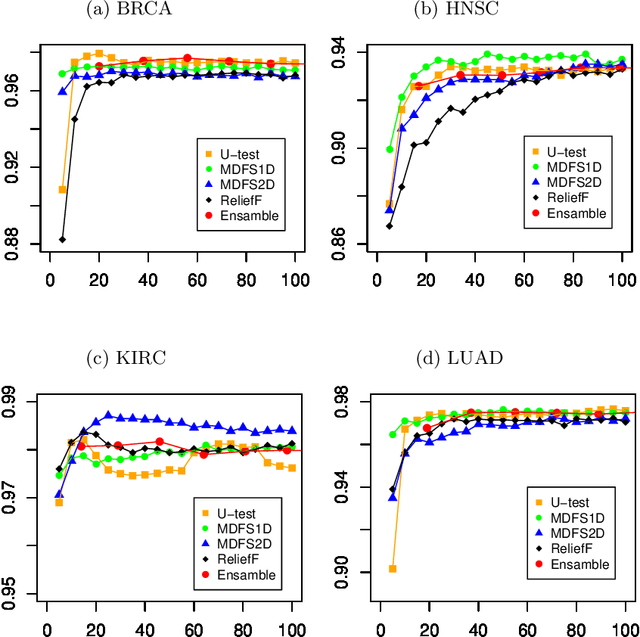

Discovery of diagnostic and prognostic molecular markers is important and actively pursued the research field in cancer research. For complex diseases, this process is often performed using Machine Learning. The current study compares two approaches for the discovery of relevant variables: by application of a single feature selection algorithm, versus by an ensemble of diverse algorithms. These approaches are used to identify variables that are relevant discerning of four cancer types using RNA-seq profiles from the Cancer Genome Atlas. The comparison is carried out in two directions: evaluating the predictive performance of models and monitoring the stability of selected variables. The most informative features are identified using a four feature selection algorithms, namely U-test, ReliefF, and two variants of the MDFS algorithm. Discerning normal and tumor tissues is performed using the Random Forest algorithm. The highest stability of the feature set was obtained when U-test was used. Unfortunately, models built on feature sets obtained from the ensemble of feature selection algorithms were no better than for models developed on feature sets obtained from individual algorithms. On the other hand, the feature selectors leading to the best classification results varied between data sets.
Bootstrap Bias Corrected Cross Validation applied to Super Learning
Mar 18, 2020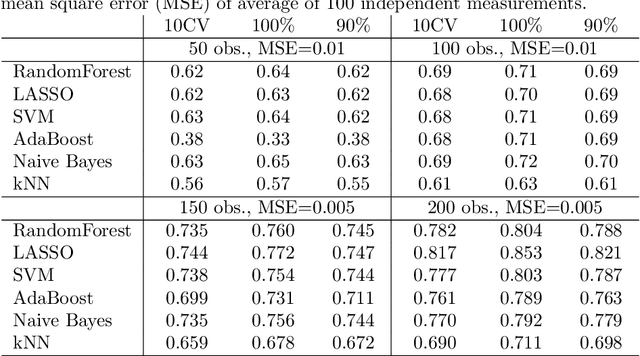

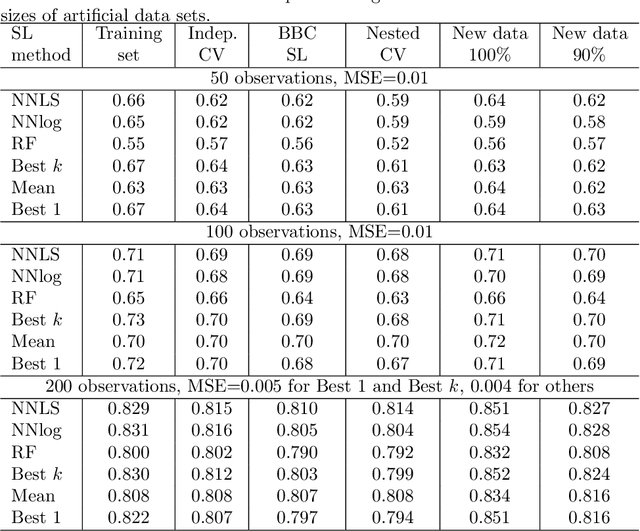

Super learner algorithm can be applied to combine results of multiple base learners to improve quality of predictions. The default method for verification of super learner results is by nested cross validation. It has been proposed by Tsamardinos et al., that nested cross validation can be replaced by resampling for tuning hyper-parameters of the learning algorithms. We apply this idea to verification of super learner and compare with other verification methods, including nested cross validation. Tests were performed on artificial data sets of diverse size and on seven real, biomedical data sets. The resampling method, called Bootstrap Bias Correction, proved to be a reasonably precise and very cost-efficient alternative for nested cross validation.
All-relevant feature selection using multidimensional filters with exhaustive search
May 16, 2017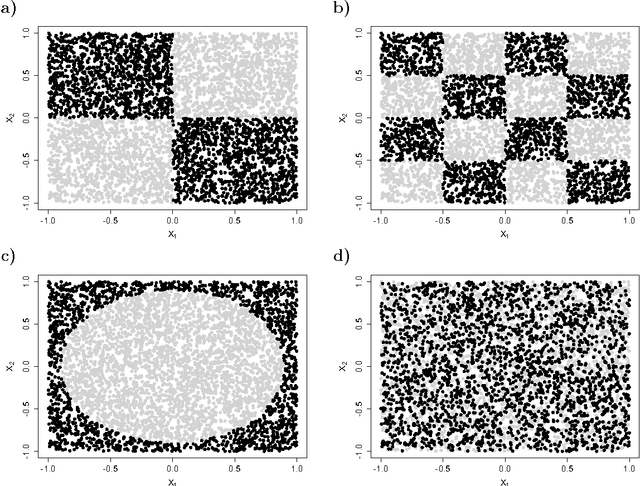
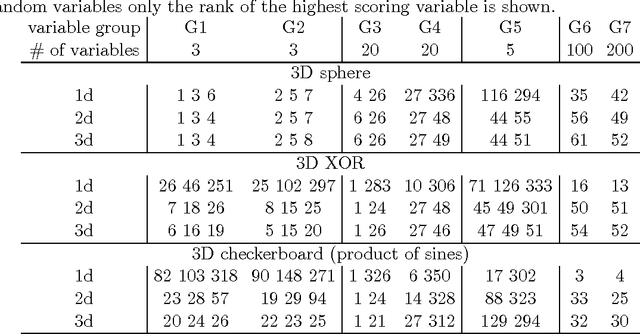


This paper describes a method for identification of the informative variables in the information system with discrete decision variables. It is targeted specifically towards discovery of the variables that are non-informative when considered alone, but are informative when the synergistic interactions between multiple variables are considered. To this end, the mutual entropy of all possible k-tuples of variables with decision variable is computed. Then, for each variable the maximal information gain due to interactions with other variables is obtained. For non-informative variables this quantity conforms to the well known statistical distributions. This allows for discerning truly informative variables from non-informative ones. For demonstration of the approach, the method is applied to several synthetic datasets that involve complex multidimensional interactions between variables. It is capable of identifying most important informative variables, even in the case when the dimensionality of the analysis is smaller than the true dimensionality of the problem. What is more, the high sensitivity of the algorithm allows for detection of the influence of nuisance variables on the response variable.
The All Relevant Feature Selection using Random Forest
Jun 25, 2011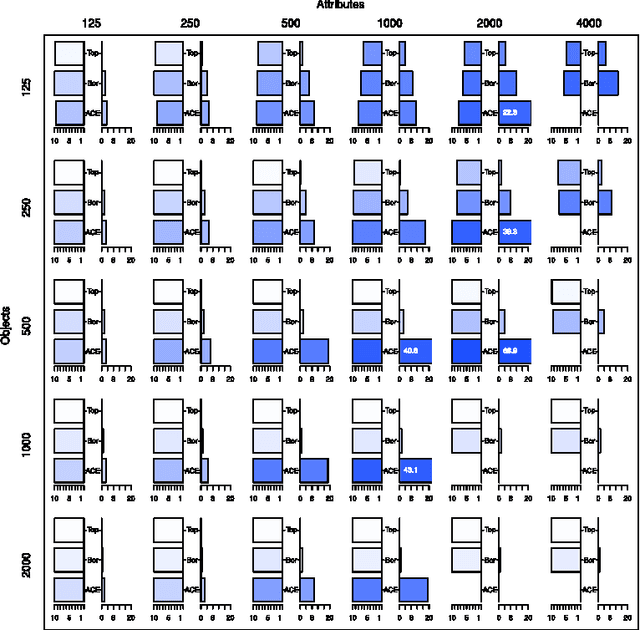
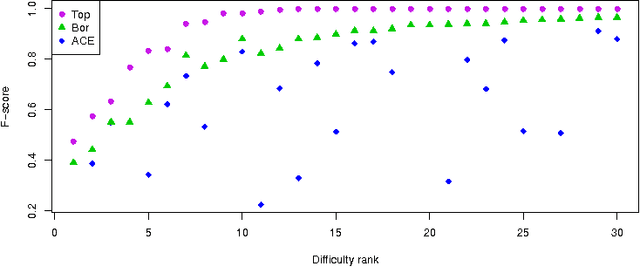
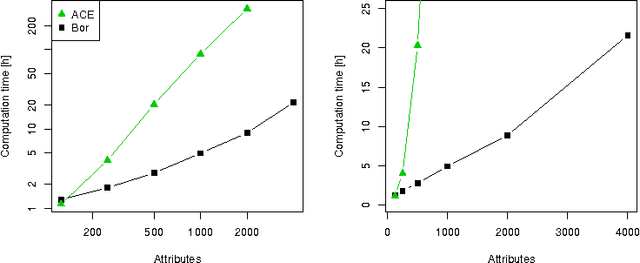
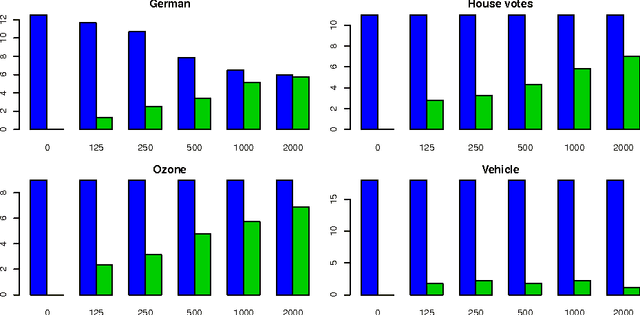
In this paper we examine the application of the random forest classifier for the all relevant feature selection problem. To this end we first examine two recently proposed all relevant feature selection algorithms, both being a random forest wrappers, on a series of synthetic data sets with varying size. We show that reasonable accuracy of predictions can be achieved and that heuristic algorithms that were designed to handle the all relevant problem, have performance that is close to that of the reference ideal algorithm. Then, we apply one of the algorithms to four families of semi-synthetic data sets to assess how the properties of particular data set influence results of feature selection. Finally we test the procedure using a well-known gene expression data set. The relevance of nearly all previously established important genes was confirmed, moreover the relevance of several new ones is discovered.
Random forest models of the retention constants in the thin layer chromatography
Jun 16, 2011

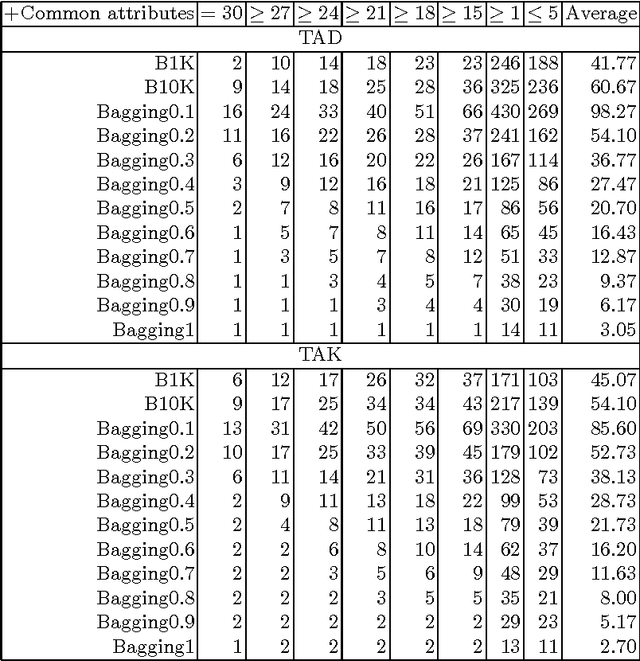
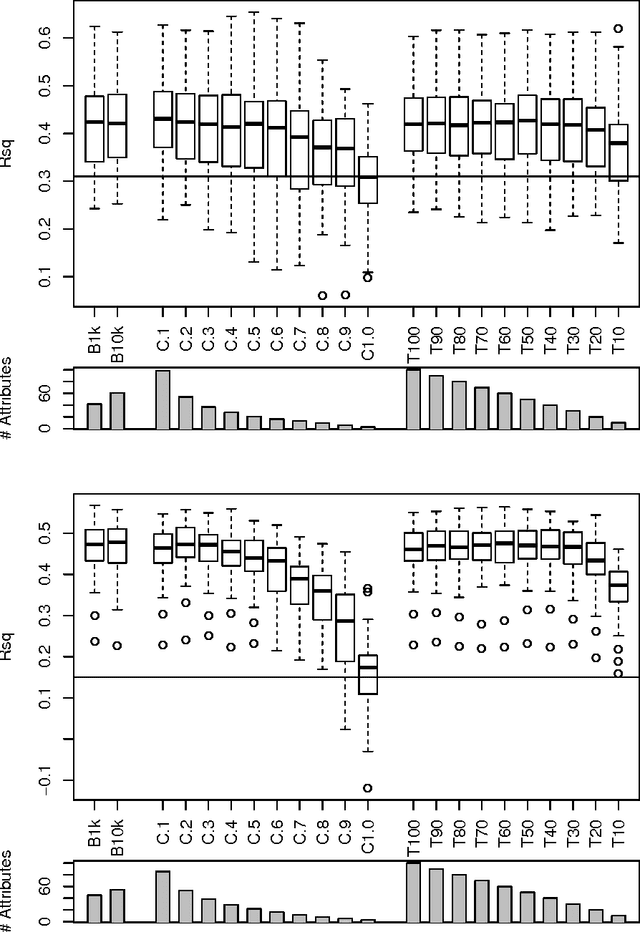
In the current study we examine an application of the machine learning methods to model the retention constants in the thin layer chromatography (TLC). This problem can be described with hundreds or even thousands of descriptors relevant to various molecular properties, most of them redundant and not relevant for the retention constant prediction. Hence we employed feature selection to significantly reduce the number of attributes. Additionally we have tested application of the bagging procedure to the feature selection. The random forest regression models were built using selected variables. The resulting models have better correlation with the experimental data than the reference models obtained with linear regression. The cross-validation confirms robustness of the models.