 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGROVER: Self-supervised Message Passing Transformer on Large-scale Molecular Data
Jun 18, 2020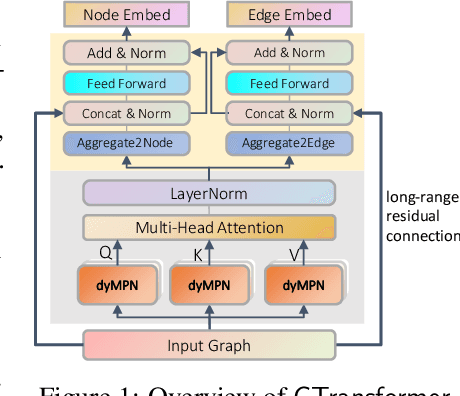


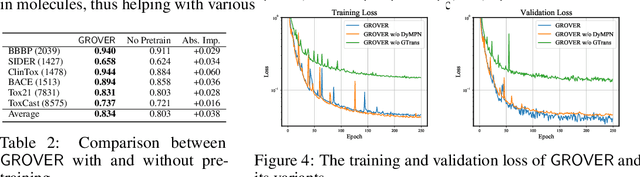
How to obtain informative representations of molecules is a crucial prerequisite in AI-driven drug design and discovery. Recent researches abstract molecules as graphs and employ Graph Neural Networks (GNNs) for task-specific and data-driven molecular representation learning. Nevertheless, two "dark clouds" impede the usage of GNNs in real scenarios: (1) insufficient labeled molecules for supervised training; (2) poor generalization capabilities to new-synthesized molecules. To address them both, we propose a novel molecular representation framework, GROVER, which stands for Graph Representation frOm self-superVised mEssage passing tRansformer. With carefully designed self-supervised tasks in node, edge and graph-level, GROVER can learn rich structural and semantic information of molecules from enormous unlabelled molecular data. Rather, to encode such complex information, GROVER integrates Message Passing Networks with the Transformer-style architecture to deliver a class of more expressive encoders of molecules. The flexibility of GROVER allows it to be trained efficiently on large-scale molecular dataset without requiring any supervision, thus being immunized to the two issues mentioned above. We pre-train GROVER with 100 million parameters on 10 million unlabelled molecules---the biggest GNN and the largest training dataset that we have ever met. We then leverage the pre-trained GROVER to downstream molecular property prediction tasks followed by task-specific fine-tuning, where we observe a huge improvement (more than 6% on average) over current state-of-the-art methods on 11 challenging benchmarks. The insights we gained are that well-designed self-supervision losses and largely-expressive pre-trained models enjoy the significant potential on performance boosting.
Multi-View Graph Neural Networks for Molecular Property Prediction
Jun 12, 2020



The crux of molecular property prediction is to generate meaningful representations of the molecules. One promising route is to exploit the molecular graph structure through Graph Neural Networks (GNNs). It is well known that both atoms and bonds significantly affect the chemical properties of a molecule, so an expressive model shall be able to exploit both node (atom) and edge (bond) information simultaneously. Guided by this observation, we present Multi-View Graph Neural Network (MV-GNN), a multi-view message passing architecture to enable more accurate predictions of molecular properties. In MV-GNN, we introduce a shared self-attentive readout component and disagreement loss to stabilize the training process. This readout component also renders the whole architecture interpretable. We further boost the expressive power of MV-GNN by proposing a cross-dependent message passing scheme that enhances information communication of the two views, which results in the MV-GNN^cross variant. Lastly, we theoretically justify the expressiveness of the two proposed models in terms of distinguishing non-isomorphism graphs. Extensive experiments demonstrate that MV-GNN models achieve remarkably superior performance over the state-of-the-art models on a variety of challenging benchmarks. Meanwhile, visualization results of the node importance are consistent with prior knowledge, which confirms the interpretability power of MV-GNN models.