 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePandemic Drugs at Pandemic Speed: Accelerating COVID-19 Drug Discovery with Hybrid Machine Learning- and Physics-based Simulations on High Performance Computers
Mar 04, 2021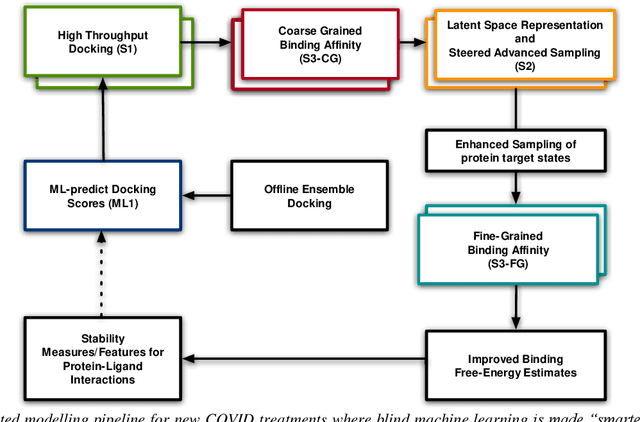

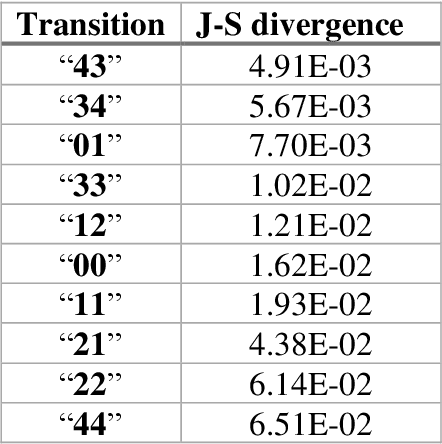
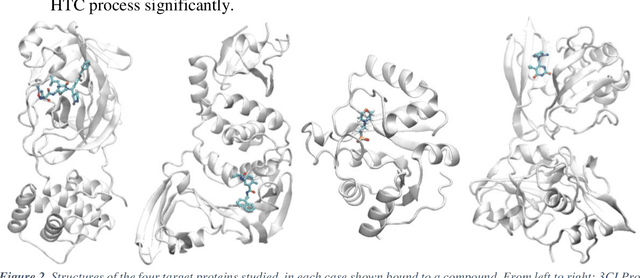
The race to meet the challenges of the global pandemic has served as a reminder that the existing drug discovery process is expensive, inefficient and slow. There is a major bottleneck screening the vast number of potential small molecules to shortlist lead compounds for antiviral drug development. New opportunities to accelerate drug discovery lie at the interface between machine learning methods, in this case developed for linear accelerators, and physics-based methods. The two in silico methods, each have their own advantages and limitations which, interestingly, complement each other. Here, we present an innovative method that combines both approaches to accelerate drug discovery. The scale of the resulting workflow is such that it is dependent on high performance computing. We have demonstrated the applicability of this workflow on four COVID-19 target proteins and our ability to perform the required large-scale calculations to identify lead compounds on a variety of supercomputers.
Distributed Kernel K-Means for Large Scale Clustering
Oct 09, 2017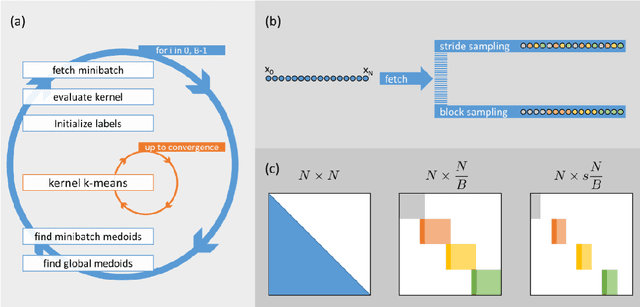
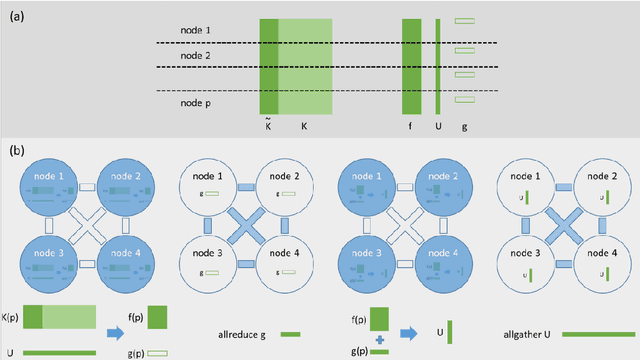
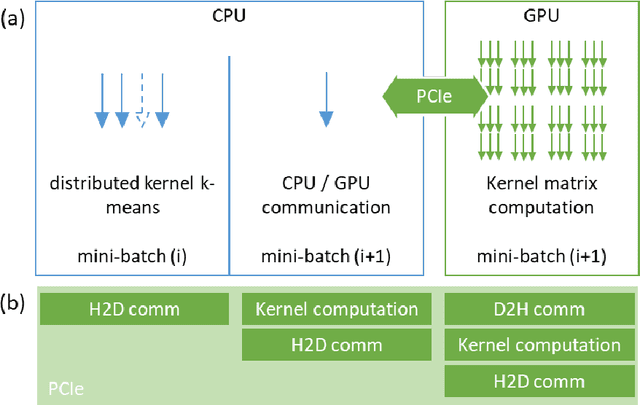
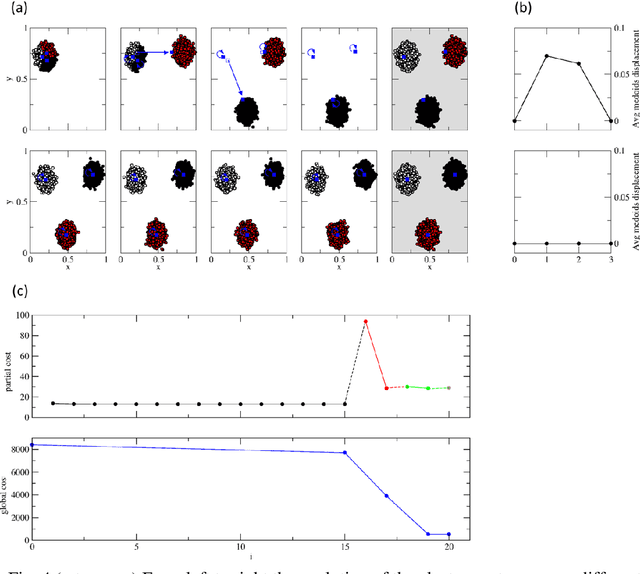
Clustering samples according to an effective metric and/or vector space representation is a challenging unsupervised learning task with a wide spectrum of applications. Among several clustering algorithms, k-means and its kernelized version have still a wide audience because of their conceptual simplicity and efficacy. However, the systematic application of the kernelized version of k-means is hampered by its inherent square scaling in memory with the number of samples. In this contribution, we devise an approximate strategy to minimize the kernel k-means cost function in which the trade-off between accuracy and velocity is automatically ruled by the available system memory. Moreover, we define an ad-hoc parallelization scheme well suited for hybrid cpu-gpu state-of-the-art parallel architectures. We proved the effectiveness both of the approximation scheme and of the parallelization method on standard UCI datasets and on molecular dynamics (MD) data in the realm of computational chemistry. In this applicative domain, clustering can play a key role for both quantitively estimating kinetics rates via Markov State Models or to give qualitatively a human compatible summarization of the underlying chemical phenomenon under study. For these reasons, we selected it as a valuable real-world application scenario.
* Conference paper