 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Canonical Correlation Analysis
Feb 03, 2025



Canonical correlation analysis (CCA) is a widely used technique for estimating associations between two sets of multi-dimensional variables. Recent advancements in CCA methods have expanded their application to decipher the interactions of multiomics datasets, imaging-omics datasets, and more. However, conventional CCA methods are limited in their ability to incorporate structured patterns in the cross-correlation matrix, potentially leading to suboptimal estimations. To address this limitation, we propose the graph Canonical Correlation Analysis (gCCA) approach, which calculates canonical correlations based on the graph structure of the cross-correlation matrix between the two sets of variables. We develop computationally efficient algorithms for gCCA, and provide theoretical results for finite sample analysis of best subset selection and canonical correlation estimation by introducing concentration inequalities and stopping time rule based on martingale theories. Extensive simulations demonstrate that gCCA outperforms competing CCA methods. Additionally, we apply gCCA to a multiomics dataset of DNA methylation and RNA-seq transcriptomics, identifying both positively and negatively regulated gene expression pathways by DNA methylation pathways.
A sparse negative binomial mixture model for clustering RNA-seq count data
Dec 05, 2019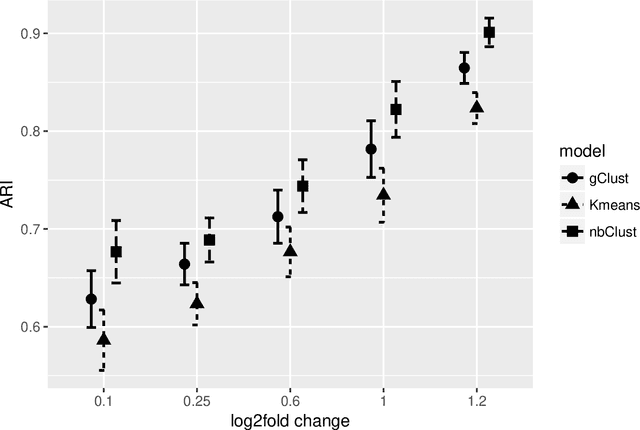
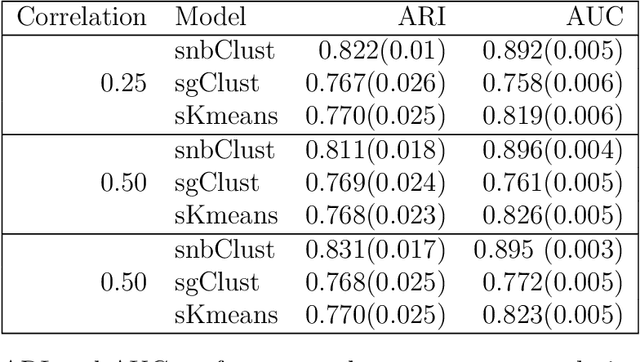
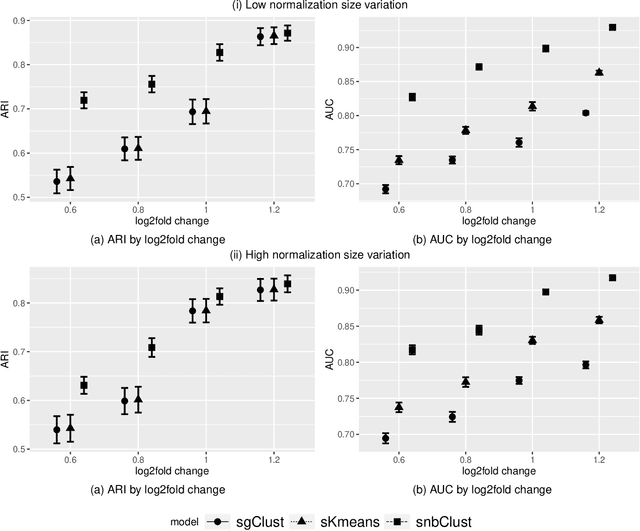

Clustering with variable selection is a challenging but critical task for modern small-n-large-p data. Existing methods based on Gaussian mixture models or sparse K-means provide solutions to continuous data. With the prevalence of RNA-seq technology and lack of count data modeling for clustering, the current practice is to normalize count expression data into continuous measures and apply existing models with Gaussian assumption. In this paper, we develop a negative binomial mixture model with gene regularization to cluster samples (small $n$) with high-dimensional gene features (large $p$). EM algorithm and Bayesian information criterion are used for inference and determining tuning parameters. The method is compared with sparse Gaussian mixture model and sparse K-means using extensive simulations and two real transcriptomic applications in breast cancer and rat brain studies. The result shows superior performance of the proposed count data model in clustering accuracy, feature selection and biological interpretation by pathway enrichment analysis.