 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCascade Detector Analysis and Application to Biomedical Microscopy
Apr 30, 2025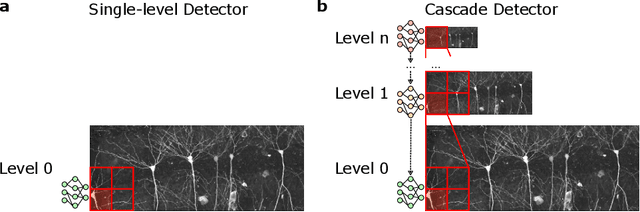
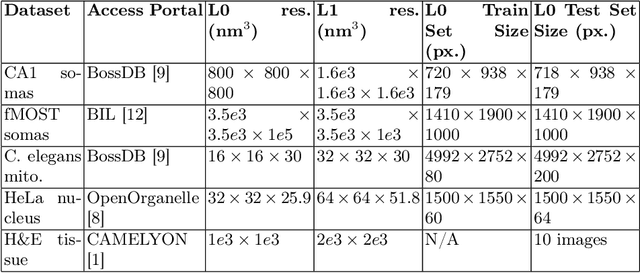
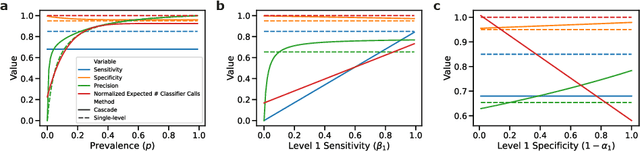

As both computer vision models and biomedical datasets grow in size, there is an increasing need for efficient inference algorithms. We utilize cascade detectors to efficiently identify sparse objects in multiresolution images. Given an object's prevalence and a set of detectors at different resolutions with known accuracies, we derive the accuracy, and expected number of classifier calls by a cascade detector. These results generalize across number of dimensions and number of cascade levels. Finally, we compare one- and two-level detectors in fluorescent cell detection, organelle segmentation, and tissue segmentation across various microscopy modalities. We show that the multi-level detector achieves comparable performance in 30-75% less time. Our work is compatible with a variety of computer vision models and data domains.
NeuroADDA: Active Discriminative Domain Adaptation in Connectomic
Mar 08, 2025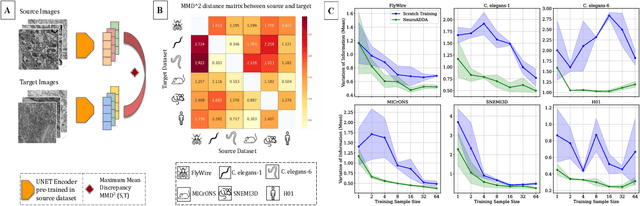


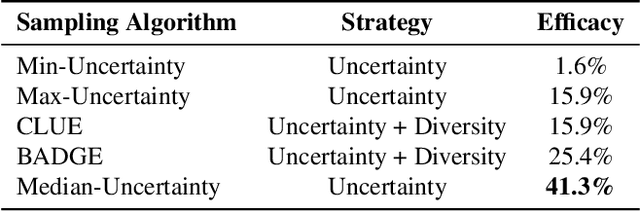
Training segmentation models from scratch has been the standard approach for new electron microscopy connectomics datasets. However, leveraging pretrained models from existing datasets could improve efficiency and performance in constrained annotation budget. In this study, we investigate domain adaptation in connectomics by analyzing six major datasets spanning different organisms. We show that, Maximum Mean Discrepancy (MMD) between neuron image distributions serves as a reliable indicator of transferability, and identifies the optimal source domain for transfer learning. Building on this, we introduce NeuroADDA, a method that combines optimal domain selection with source-free active learning to effectively adapt pretrained backbones to a new dataset. NeuroADDA consistently outperforms training from scratch across diverse datasets and fine-tuning sample sizes, with the largest gain observed at $n=4$ samples with a 25-67\% reduction in Variation of Information. Finally, we show that our analysis of distributional differences among neuron images from multiple species in a learned feature space reveals that these domain "distances" correlate with phylogenetic distance among those species.
Hidden Markov Modeling for Maximum Likelihood Neuron Reconstruction
Jun 04, 2021

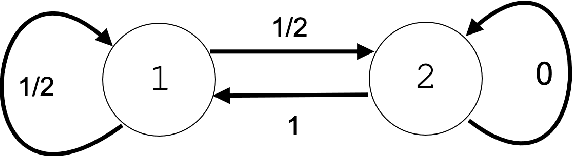

Recent advances in brain clearing and imaging have made it possible to image entire mammalian brains at sub-micron resolution. These images offer the potential to assemble brain-wide atlases of projection neuron morphology, but manual neuron reconstruction remains a bottleneck. Here we present a method inspired by hidden Markov modeling and appearance modeling of fluorescent neuron images that can automatically trace neuronal processes. Our method leverages dynamic programming to scale to terabyte sized image data and can be applied to images with one or more neurons. We applied our algorithm to the output of image segmentation models where false negatives severed neuronal processes, and showed that it can follow axons in the presence of noise or nearby neurons. Our method has the potential to be integrated into a semi or fully automated reconstruction pipeline. Additionally, it creates a framework through which users can intervene with hard constraints to, for example, rule out certain reconstructions, or assign axons to particular cell bodies.
AutoGMM: Automatic Gaussian Mixture Modeling in Python
Oct 04, 2019
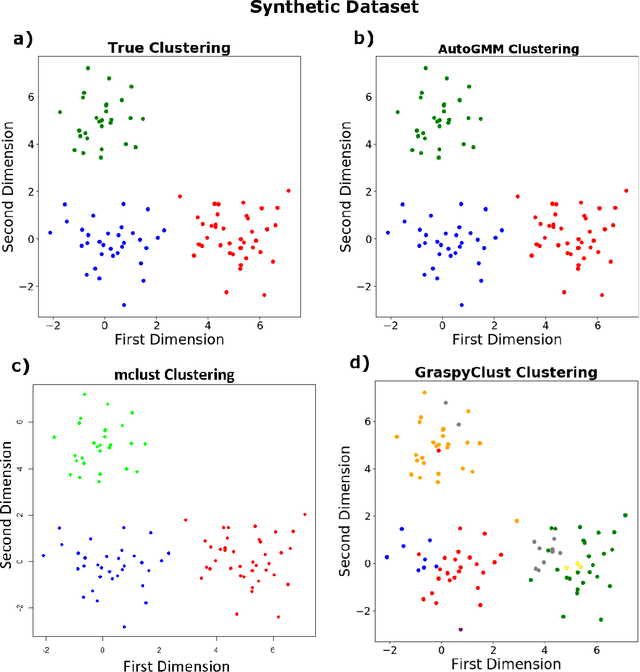
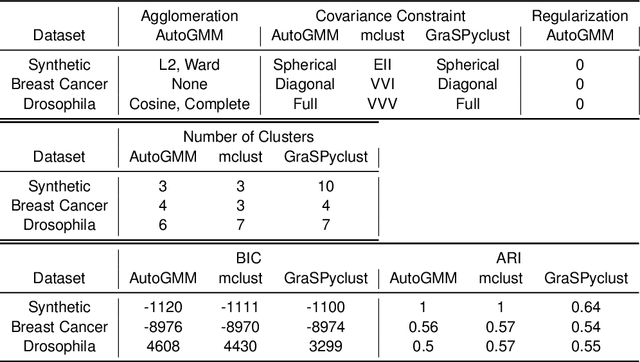
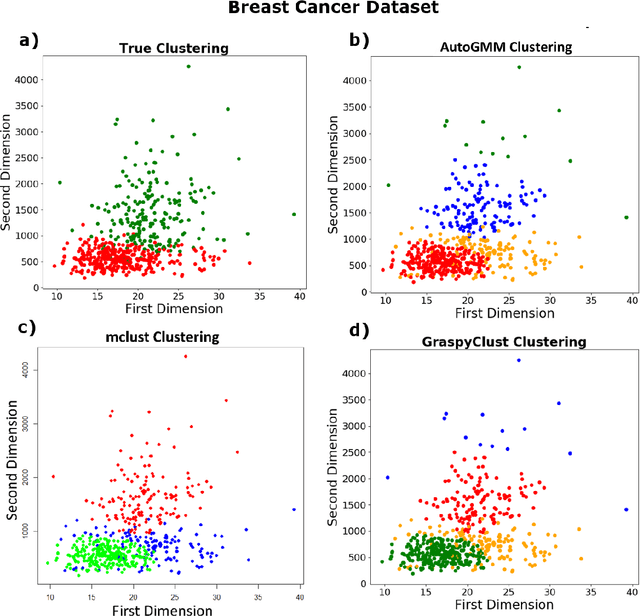
Gaussian mixture modeling is a fundamental tool in clustering, as well as discriminant analysis and semiparametric density estimation. However, estimating the optimal model for any given number of components is an NP-hard problem, and estimating the number of components is in some respects an even harder problem. In R, a popular package called mclust addresses both of these problems. However, Python has lacked such a package. We therefore introduce AutoGMM, a Python algorithm for automatic Gaussian mixture modeling. AutoGMM builds upon scikit-learn's AgglomerativeClustering and GaussianMixture classes, with certain modifications to make the results more stable. Empirically, on several different applications, AutoGMM performs approximately as well as mclust. This algorithm is freely available and therefore further shrinks the gap between functionality of R and Python for data science.