 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssessing Large Language Models for Medical QA: Zero-Shot and LLM-as-a-Judge Evaluation
Feb 16, 2026Recently, Large Language Models (LLMs) have gained significant traction in medical domain, especially in developing a QA systems to Medical QA systems for enhancing access to healthcare in low-resourced settings. This paper compares five LLMs deployed between April 2024 and August 2025 for medical QA, using the iCliniq dataset, containing 38,000 medical questions and answers of diverse specialties. Our models include Llama-3-8B-Instruct, Llama 3.2 3B, Llama 3.3 70B Instruct, Llama-4-Maverick-17B-128E-Instruct, and GPT-5-mini. We are using a zero-shot evaluation methodology and using BLEU and ROUGE metrics to evaluate performance without specialized fine-tuning. Our results show that larger models like Llama 3.3 70B Instruct outperform smaller models, consistent with observed scaling benefits in clinical tasks. It is notable that, Llama-4-Maverick-17B exhibited more competitive results, thus highlighting evasion efficiency trade-offs relevant for practical deployment. These findings align with advancements in LLM capabilities toward professional-level medical reasoning and reflect the increasing feasibility of LLM-supported QA systems in the real clinical environments. This benchmark aims to serve as a standardized setting for future study to minimize model size, computational resources and to maximize clinical utility in medical NLP applications.
Faithful Summarization of Consumer Health Queries: A Cross-Lingual Framework with LLMs
Nov 13, 2025Summarizing consumer health questions (CHQs) can ease communication in healthcare, but unfaithful summaries that misrepresent medical details pose serious risks. We propose a framework that combines TextRank-based sentence extraction and medical named entity recognition with large language models (LLMs) to enhance faithfulness in medical text summarization. In our experiments, we fine-tuned the LLaMA-2-7B model on the MeQSum (English) and BanglaCHQ-Summ (Bangla) datasets, achieving consistent improvements across quality (ROUGE, BERTScore, readability) and faithfulness (SummaC, AlignScore) metrics, and outperforming zero-shot baselines and prior systems. Human evaluation further shows that over 80\% of generated summaries preserve critical medical information. These results highlight faithfulness as an essential dimension for reliable medical summarization and demonstrate the potential of our approach for safer deployment of LLMs in healthcare contexts.
BanglaMedQA and BanglaMMedBench: Evaluating Retrieval-Augmented Generation Strategies for Bangla Biomedical Question Answering
Nov 06, 2025Developing accurate biomedical Question Answering (QA) systems in low-resource languages remains a major challenge, limiting equitable access to reliable medical knowledge. This paper introduces BanglaMedQA and BanglaMMedBench, the first large-scale Bangla biomedical Multiple Choice Question (MCQ) datasets designed to evaluate reasoning and retrieval in medical artificial intelligence (AI). The study applies and benchmarks several Retrieval-Augmented Generation (RAG) strategies, including Traditional, Zero-Shot Fallback, Agentic, Iterative Feedback, and Aggregate RAG, combining textbook-based and web retrieval with generative reasoning to improve factual accuracy. A key novelty lies in integrating a Bangla medical textbook corpus through Optical Character Recognition (OCR) and implementing an Agentic RAG pipeline that dynamically selects between retrieval and reasoning strategies. Experimental results show that the Agentic RAG achieved the highest accuracy 89.54% with openai/gpt-oss-120b, outperforming other configurations and demonstrating superior rationale quality. These findings highlight the potential of RAG-based methods to enhance the reliability and accessibility of Bangla medical QA, establishing a foundation for future research in multilingual medical artificial intelligence.
A Pan-cancer Classification Model using Multi-view Feature Selection Method and Ensemble Classifier
Jan 12, 2025



Accurately identifying cancer samples is crucial for precise diagnosis and effective patient treatment. Traditional methods falter with high-dimensional and high feature-to-sample count ratios, which are critical for classifying cancer samples. This study aims to develop a novel feature selection framework specifically for transcriptome data and propose two ensemble classifiers. For feature selection, we partition the transcriptome dataset vertically based on feature types. Then apply the Boruta feature selection process on each of the partitions, combine the results, and apply Boruta again on the combined result. We repeat the process with different parameters of Boruta and prepare the final feature set. Finally, we constructed two ensemble ML models based on LR, SVM and XGBoost classifiers with max voting and averaging probability approach. We used 10-fold cross-validation to ensure robust and reliable classification performance. With 97.11\% accuracy and 0.9996 AUC value, our approach performs better compared to existing state-of-the-art methods to classify 33 types of cancers. A set of 12 types of cancer is traditionally challenging to differentiate between each other due to their similarity in tissue of origin. Our method accurately identifies over 90\% of samples from these 12 types of cancers, which outperforms all known methods presented in existing literature. The gene set enrichment analysis reveals that our framework's selected features have enriched the pathways highly related to cancers. This study develops a feature selection framework to select features highly related to cancer development and leads to identifying different types of cancer samples with higher accuracy.
Precision Cancer Classification and Biomarker Identification from mRNA Gene Expression via Dimensionality Reduction and Explainable AI
Oct 08, 2024Gene expression analysis is a critical method for cancer classification, enabling precise diagnoses through the identification of unique molecular signatures associated with various tumors. Identifying cancer-specific genes from gene expression values enables a more tailored and personalized treatment approach. However, the high dimensionality of mRNA gene expression data poses challenges for analysis and data extraction. This research presents a comprehensive pipeline designed to accurately identify 33 distinct cancer types and their corresponding gene sets. It incorporates a combination of normalization and feature selection techniques to reduce dataset dimensionality effectively while ensuring high performance. Notably, our pipeline successfully identifies a substantial number of cancer-specific genes using a reduced feature set of just 500, in contrast to using the full dataset comprising 19,238 features. By employing an ensemble approach that combines three top-performing classifiers, a classification accuracy of 96.61% was achieved. Furthermore, we leverage Explainable AI to elucidate the biological significance of the identified cancer-specific genes, employing Differential Gene Expression (DGE) analysis.
Nuclei Instance Segmentation of Cryosectioned H&E Stained Histological Images using Triple U-Net Architecture
Apr 19, 2024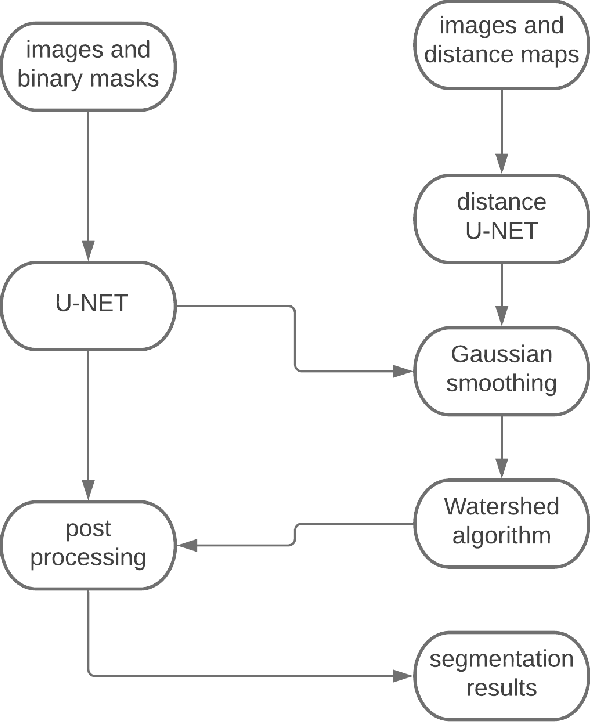

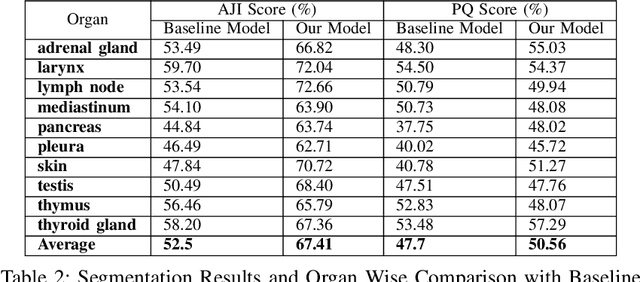
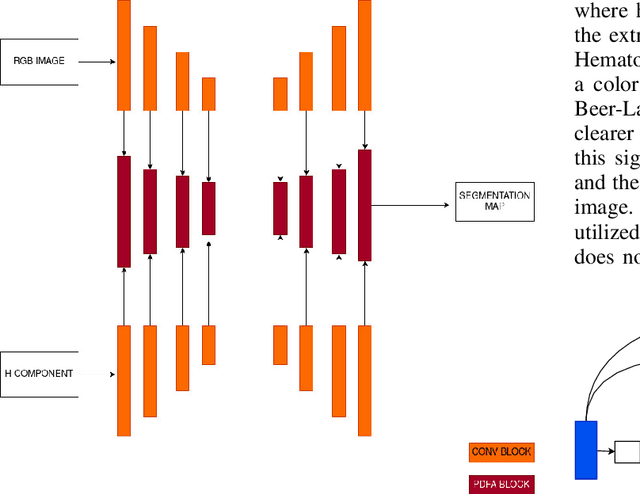
Nuclei instance segmentation is crucial in oncological diagnosis and cancer pathology research. H&E stained images are commonly used for medical diagnosis, but pre-processing is necessary before using them for image processing tasks. Two principal pre-processing methods are formalin-fixed paraffin-embedded samples (FFPE) and frozen tissue samples (FS). While FFPE is widely used, it is time-consuming, while FS samples can be processed quickly. Analyzing H&E stained images derived from fast sample preparation, staining, and scanning can pose difficulties due to the swift process, which can result in the degradation of image quality. This paper proposes a method that leverages the unique optical characteristics of H&E stained images. A three-branch U-Net architecture has been implemented, where each branch contributes to the final segmentation results. The process includes applying watershed algorithm to separate overlapping regions and enhance accuracy. The Triple U-Net architecture comprises an RGB branch, a Hematoxylin branch, and a Segmentation branch. This study focuses on a novel dataset named CryoNuSeg. The results obtained through robust experiments outperform the state-of-the-art results across various metrics. The benchmark score for this dataset is AJI 52.5 and PQ 47.7, achieved through the implementation of U-Net Architecture. However, the proposed Triple U-Net architecture achieves an AJI score of 67.41 and PQ of 50.56. The proposed architecture improves more on AJI than other evaluation metrics, which further justifies the superiority of the Triple U-Net architecture over the baseline U-Net model, as AJI is a more strict evaluation metric. The use of the three-branch U-Net model, followed by watershed post-processing, significantly surpasses the benchmark scores, showing substantial improvement in the AJI score