 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Artificial Scientist -- in-transit Machine Learning of Plasma Simulations
Jan 06, 2025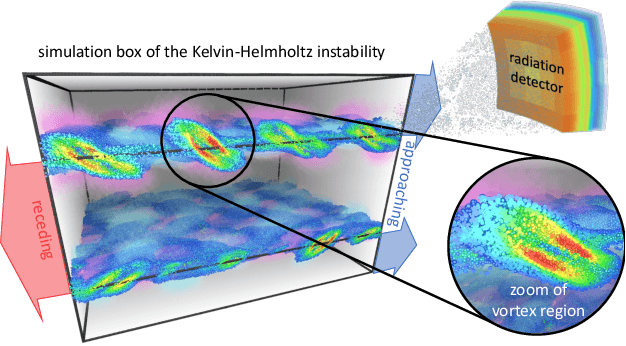
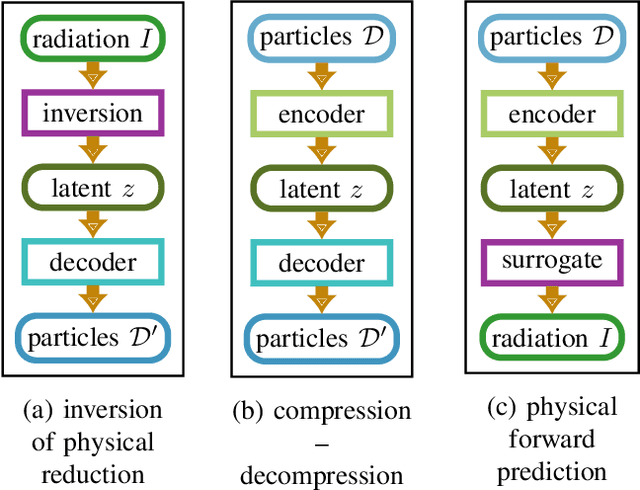
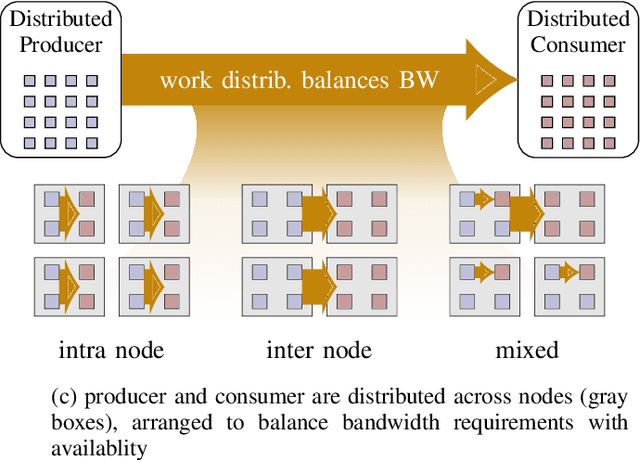
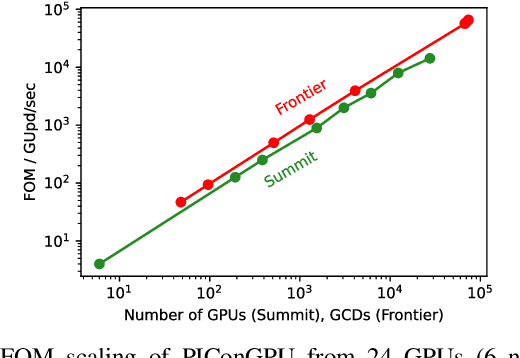
Increasing HPC cluster sizes and large-scale simulations that produce petabytes of data per run, create massive IO and storage challenges for analysis. Deep learning-based techniques, in particular, make use of these amounts of domain data to extract patterns that help build scientific understanding. Here, we demonstrate a streaming workflow in which simulation data is streamed directly to a machine-learning (ML) framework, circumventing the file system bottleneck. Data is transformed in transit, asynchronously to the simulation and the training of the model. With the presented workflow, data operations can be performed in common and easy-to-use programming languages, freeing the application user from adapting the application output routines. As a proof-of-concept we consider a GPU accelerated particle-in-cell (PIConGPU) simulation of the Kelvin- Helmholtz instability (KHI). We employ experience replay to avoid catastrophic forgetting in learning from this non-steady process in a continual manner. We detail challenges addressed while porting and scaling to Frontier exascale system.
Materials Learning Algorithms (MALA): Scalable Machine Learning for Electronic Structure Calculations in Large-Scale Atomistic Simulations
Nov 29, 2024



We present the Materials Learning Algorithms (MALA) package, a scalable machine learning framework designed to accelerate density functional theory (DFT) calculations suitable for large-scale atomistic simulations. Using local descriptors of the atomic environment, MALA models efficiently predict key electronic observables, including local density of states, electronic density, density of states, and total energy. The package integrates data sampling, model training and scalable inference into a unified library, while ensuring compatibility with standard DFT and molecular dynamics codes. We demonstrate MALA's capabilities with examples including boron clusters, aluminum across its solid-liquid phase boundary, and predicting the electronic structure of a stacking fault in a large beryllium slab. Scaling analyses reveal MALA's computational efficiency and identify bottlenecks for future optimization. With its ability to model electronic structures at scales far beyond standard DFT, MALA is well suited for modeling complex material systems, making it a versatile tool for advanced materials research.
Machine Learning State-of-the-Art with Uncertainties
Apr 14, 2022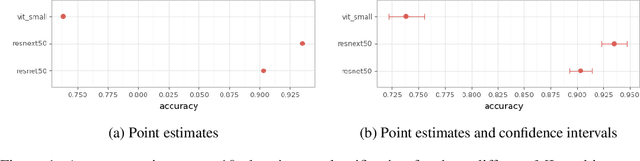
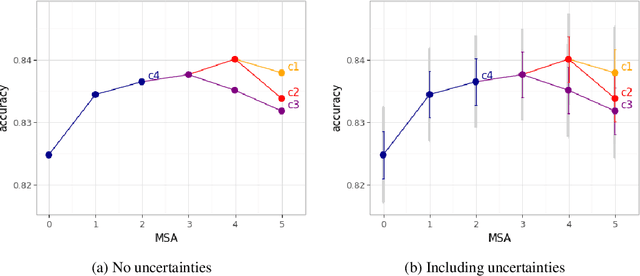
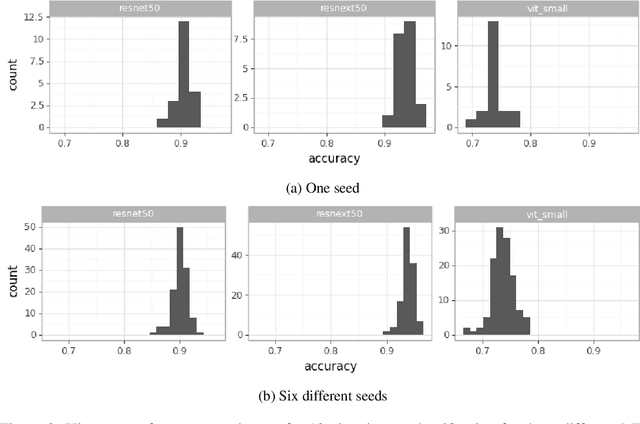
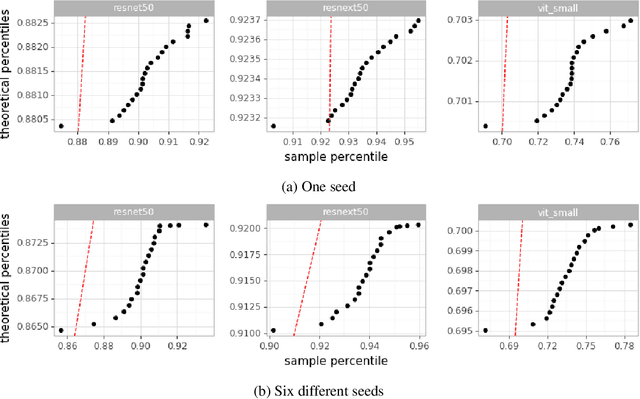
With the availability of data, hardware, software ecosystem and relevant skill sets, the machine learning community is undergoing a rapid development with new architectures and approaches appearing at high frequency every year. In this article, we conduct an exemplary image classification study in order to demonstrate how confidence intervals around accuracy measurements can greatly enhance the communication of research results as well as impact the reviewing process. In addition, we explore the hallmarks and limitations of this approximation. We discuss the relevance of this approach reflecting on a spotlight publication of ICLR22. A reproducible workflow is made available as an open-source adjoint to this publication. Based on our discussion, we make suggestions for improving the authoring and reviewing process of machine learning articles.