 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient Symmetry-Aware Materials Generation via Hierarchical Generative Flow Networks
Nov 06, 2024



Discovering new solid-state materials requires rapidly exploring the vast space of crystal structures and locating stable regions. Generating stable materials with desired properties and compositions is extremely difficult as we search for very small isolated pockets in the exponentially many possibilities, considering elements from the periodic table and their 3D arrangements in crystal lattices. Materials discovery necessitates both optimized solution structures and diversity in the generated material structures. Existing methods struggle to explore large material spaces and generate diverse samples with desired properties and requirements. We propose the Symmetry-aware Hierarchical Architecture for Flow-based Traversal (SHAFT), a novel generative model employing a hierarchical exploration strategy to efficiently exploit the symmetry of the materials space to generate crystal structures given desired properties. In particular, our model decomposes the exponentially large materials space into a hierarchy of subspaces consisting of symmetric space groups, lattice parameters, and atoms. We demonstrate that SHAFT significantly outperforms state-of-the-art iterative generative methods, such as Generative Flow Networks (GFlowNets) and Crystal Diffusion Variational AutoEncoders (CDVAE), in crystal structure generation tasks, achieving higher validity, diversity, and stability of generated structures optimized for target properties and requirements.
Naturally-meaningful and efficient descriptors: machine learning of material properties based on robust one-shot ab initio descriptors
Mar 02, 2022
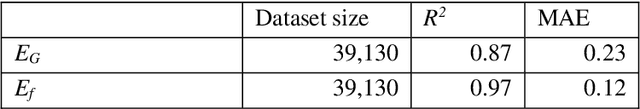


Establishing a data-driven pipeline for the discovery of novel materials requires the engineering of material features that can be feasibly calculated and can be applied to predict a material's target properties. Here we propose a new class of descriptors for describing crystal structures, which we term Robust One-Shot Ab initio (ROSA) descriptors. ROSA is computationally cheap and is shown to accurately predict a range of material properties. These simple and intuitive class of descriptors are generated from the energetics of a material at a low level of theory using an incomplete ab initio calculation. We demonstrate how the incorporation of ROSA descriptors in ML-based property prediction leads to accurate predictions over a wide range of crystals, amorphized crystals, metal-organic frameworks and molecules. We believe that the low computational cost and ease of use of these descriptors will significantly improve ML-based predictions.