 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSketch-Augmented Features Improve Learning Long-Range Dependencies in Graph Neural Networks
Nov 05, 2025Graph Neural Networks learn on graph-structured data by iteratively aggregating local neighborhood information. While this local message passing paradigm imparts a powerful inductive bias and exploits graph sparsity, it also yields three key challenges: (i) oversquashing of long-range information, (ii) oversmoothing of node representations, and (iii) limited expressive power. In this work we inject randomized global embeddings of node features, which we term \textit{Sketched Random Features}, into standard GNNs, enabling them to efficiently capture long-range dependencies. The embeddings are unique, distance-sensitive, and topology-agnostic -- properties which we analytically and empirically show alleviate the aforementioned limitations when injected into GNNs. Experimental results on real-world graph learning tasks confirm that this strategy consistently improves performance over baseline GNNs, offering both a standalone solution and a complementary enhancement to existing techniques such as graph positional encodings. Our source code is available at \href{https://github.com/ryienh/sketched-random-features}{https://github.com/ryienh/sketched-random-features}.
Quality Measures for Dynamic Graph Generative Models
Mar 03, 2025Deep generative models have recently achieved significant success in modeling graph data, including dynamic graphs, where topology and features evolve over time. However, unlike in vision and natural language domains, evaluating generative models for dynamic graphs is challenging due to the difficulty of visualizing their output, making quantitative metrics essential. In this work, we develop a new quality metric for evaluating generative models of dynamic graphs. Current metrics for dynamic graphs typically involve discretizing the continuous-evolution of graphs into static snapshots and then applying conventional graph similarity measures. This approach has several limitations: (a) it models temporally related events as i.i.d. samples, failing to capture the non-uniform evolution of dynamic graphs; (b) it lacks a unified measure that is sensitive to both features and topology; (c) it fails to provide a scalar metric, requiring multiple metrics without clear superiority; and (d) it requires explicitly instantiating each static snapshot, leading to impractical runtime demands that hinder evaluation at scale. We propose a novel metric based on the \textit{Johnson-Lindenstrauss} lemma, applying random projections directly to dynamic graph data. This results in an expressive, scalar, and application-agnostic measure of dynamic graph similarity that overcomes the limitations of traditional methods. We also provide a comprehensive empirical evaluation of metrics for continuous-time dynamic graphs, demonstrating the effectiveness of our approach compared to existing methods. Our implementation is available at https://github.com/ryienh/jl-metric.
A Deep Probabilistic Framework for Continuous Time Dynamic Graph Generation
Dec 20, 2024



Recent advancements in graph representation learning have shifted attention towards dynamic graphs, which exhibit evolving topologies and features over time. The increased use of such graphs creates a paramount need for generative models suitable for applications such as data augmentation, obfuscation, and anomaly detection. However, there are few generative techniques that handle continuously changing temporal graph data; existing work largely relies on augmenting static graphs with additional temporal information to model dynamic interactions between nodes. In this work, we propose a fundamentally different approach: We instead directly model interactions as a joint probability of an edge forming between two nodes at a given time. This allows us to autoregressively generate new synthetic dynamic graphs in a largely assumption free, scalable, and inductive manner. We formalize this approach as DG-Gen, a generative framework for continuous time dynamic graphs, and demonstrate its effectiveness over five datasets. Our experiments demonstrate that DG-Gen not only generates higher fidelity graphs compared to traditional methods but also significantly advances link prediction tasks.
Deep Surrogate Docking: Accelerating Automated Drug Discovery with Graph Neural Networks
Nov 04, 2022



The process of screening molecules for desirable properties is a key step in several applications, ranging from drug discovery to material design. During the process of drug discovery specifically, protein-ligand docking, or chemical docking, is a standard in-silico scoring technique that estimates the binding affinity of molecules with a specific protein target. Recently, however, as the number of virtual molecules available to test has rapidly grown, these classical docking algorithms have created a significant computational bottleneck. We address this problem by introducing Deep Surrogate Docking (DSD), a framework that applies deep learning-based surrogate modeling to accelerate the docking process substantially. DSD can be interpreted as a formalism of several earlier surrogate prefiltering techniques, adding novel metrics and practical training practices. Specifically, we show that graph neural networks (GNNs) can serve as fast and accurate estimators of classical docking algorithms. Additionally, we introduce FiLMv2, a novel GNN architecture which we show outperforms existing state-of-the-art GNN architectures, attaining more accurate and stable performance by allowing the model to filter out irrelevant information from data more efficiently. Through extensive experimentation and analysis, we show that the DSD workflow combined with the FiLMv2 architecture provides a 9.496x speedup in molecule screening with a <3% recall error rate on an example docking task. Our open-source code is available at https://github.com/ryienh/graph-dock.
Operation-Level Performance Benchmarking of Graph Neural Networks for Scientific Applications
Jul 20, 2022

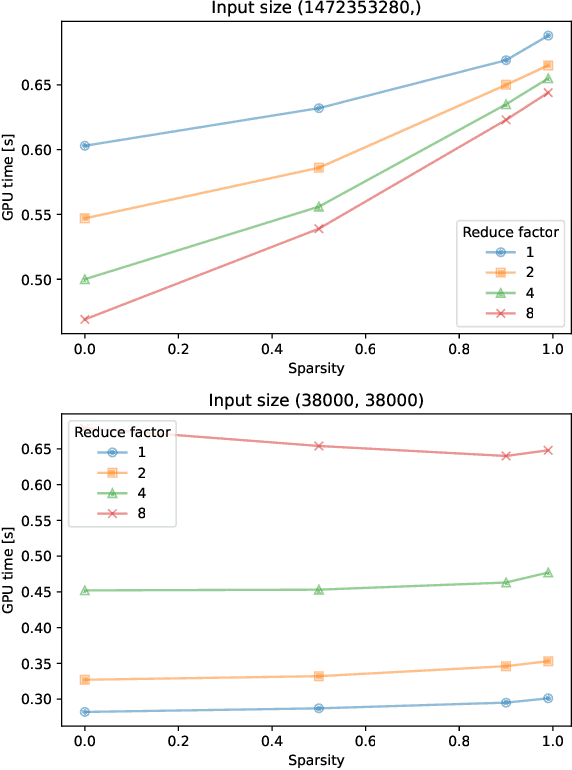
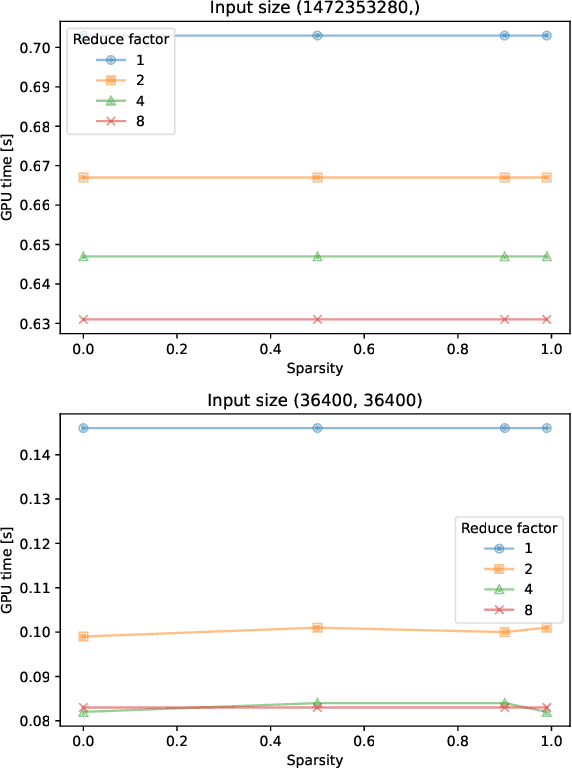
As Graph Neural Networks (GNNs) increase in popularity for scientific machine learning, their training and inference efficiency is becoming increasingly critical. Additionally, the deep learning field as a whole is trending towards wider and deeper networks, and ever increasing data sizes, to the point where hard hardware bottlenecks are often encountered. Emerging specialty hardware platforms provide an exciting solution to this problem. In this paper, we systematically profile and select low-level operations pertinent to GNNs for scientific computing implemented in the Pytorch Geometric software framework. These are then rigorously benchmarked on NVIDIA A100 GPUs for several various combinations of input values, including tensor sparsity. We then analyze these results for each operation. At a high level, we conclude that on NVIDIA systems: (1) confounding bottlenecks such as memory inefficiency often dominate runtime costs moreso than data sparsity alone, (2) native Pytorch operations are often as or more competitive than their Pytorch Geometric equivalents, especially at low to moderate levels of input data sparsity, and (3) many operations central to state-of-the-art GNN architectures have little to no optimization for sparsity. We hope that these results serve as a baseline for those developing these operations on specialized hardware and that our subsequent analysis helps to facilitate future software and hardware based optimizations of these operations and thus scalable GNN performance as a whole.