 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnity Smoothing for Handling Inconsistent Evidence in Bayesian Networks and Unity Propagation for Faster Inference
Jan 21, 2022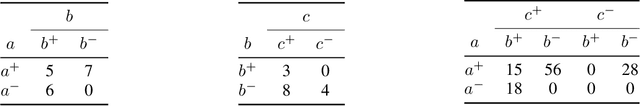
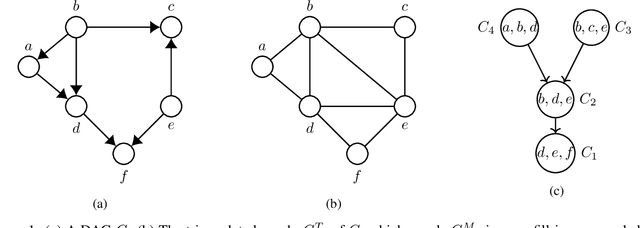

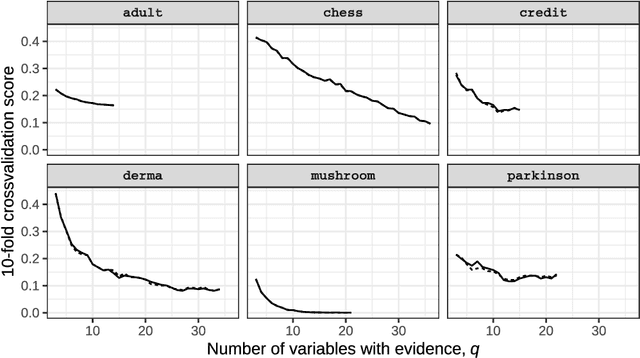
We propose Unity Smoothing (US) for handling inconsistencies between a Bayesian network model and new unseen observations. We show that prediction accuracy, using the junction tree algorithm with US is comparable to that of Laplace smoothing. Moreover, in applications were sparsity of the data structures is utilized, US outperforms Laplace smoothing in terms of memory usage. Furthermore, we detail how to avoid redundant calculations that must otherwise be performed during the message passing scheme in the junction tree algorithm which we refer to as Unity Propagation (UP). Experimental results shows that it is always faster to exploit UP on top of the Lauritzen-Spigelhalter message passing scheme for the junction tree algorithm.
DNA mixture deconvolution using an evolutionary algorithm with multiple populations, hill-climbing, and guided mutation
Dec 01, 2020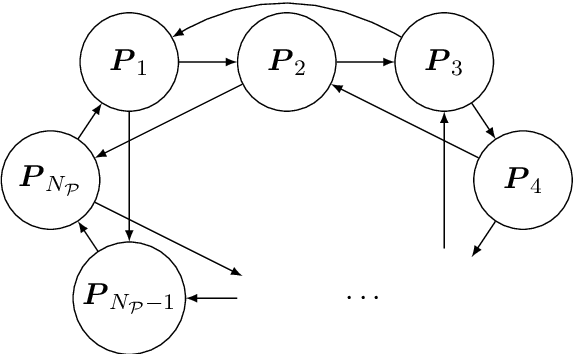



DNA samples crime cases analysed in forensic genetics, frequently contain DNA from multiple contributors. These occur as convolutions of the DNA profiles of the individual contributors to the DNA sample. Thus, in cases where one or more of the contributors were unknown, an objective of interest would be the separation, often called deconvolution, of these unknown profiles. In order to obtain deconvolutions of the unknown DNA profiles, we introduced a multiple population evolutionary algorithm (MEA). We allowed the mutation operator of the MEA to utilise that the fitness is based on a probabilistic model and guide it by using the deviations between the observed and the expected value for every element of the encoded individual. This guided mutation operator (GM) was designed such that the larger the deviation the higher probability of mutation. Furthermore, the GM was inhomogeneous in time, decreasing to a specified lower bound as the number of iterations increased. We analysed 102 two-person DNA mixture samples in varying mixture proportions. The samples were quantified using two different DNA prep. kits: (1) Illumina ForenSeq Panel B (30 samples), and (2) Applied Biosystems Precision ID Globalfiler NGS STR panel (72 samples). The DNA mixtures were deconvoluted by the MEA and compared to the true DNA profiles of the sample. We analysed three scenarios where we assumed: (1) the DNA profile of the major contributor was unknown, (2) DNA profile of the minor was unknown, and (3) both DNA profiles were unknown. Furthermore, we conducted a series of sensitivity experiments on the ForenSeq panel by varying the sub-population size, comparing a completely random homogeneous mutation operator to the guided operator with varying mutation decay rates, and allowing for hill-climbing of the parent population.
Estimating a common covariance matrix for network meta-analysis of gene expression datasets in diffuse large B-cell lymphoma
Aug 21, 2017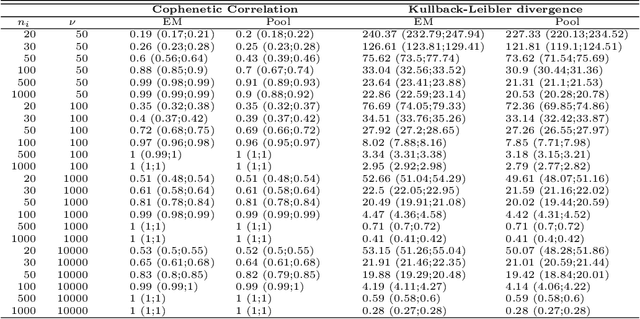

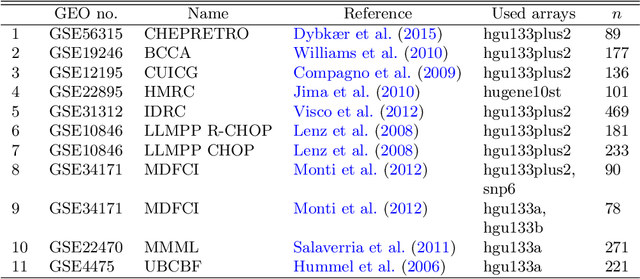
The estimation of covariance matrices of gene expressions has many applications in cancer systems biology. Many gene expression studies, however, are hampered by low sample size and it has therefore become popular to increase sample size by collecting gene expression data across studies. Motivated by the traditional meta-analysis using random effects models, we present a hierarchical random covariance model and use it for the meta-analysis of gene correlation networks across 11 large-scale gene expression studies of diffuse large B-cell lymphoma (DLBCL). We suggest to use a maximum likelihood estimator for the underlying common covariance matrix and introduce an EM algorithm for estimation. By simulation experiments comparing the estimated covariance matrices by cophenetic correlation and Kullback-Leibler divergence the suggested estimator showed to perform better or not worse than a simple pooled estimator. In a posthoc analysis of the estimated common covariance matrix for the DLBCL data we were able to identify novel biologically meaningful gene correlation networks with eigengenes of prognostic value. In conclusion, the method seems to provide a generally applicable framework for meta-analysis, when multiple features are measured and believed to share a common covariance matrix obscured by study dependent noise.
Targeted Fused Ridge Estimation of Inverse Covariance Matrices from Multiple High-Dimensional Data Classes
Sep 26, 2015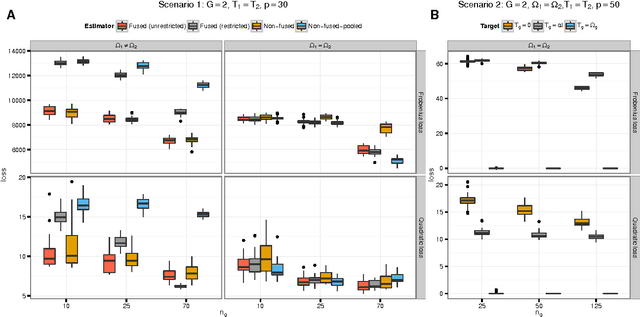
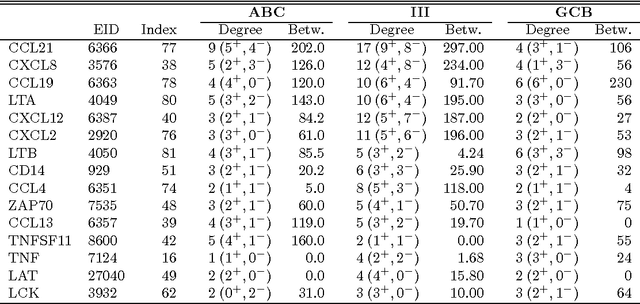
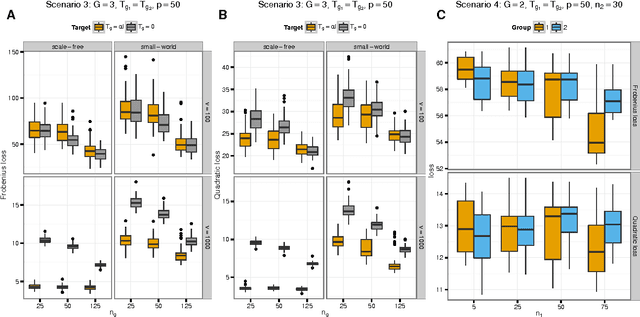
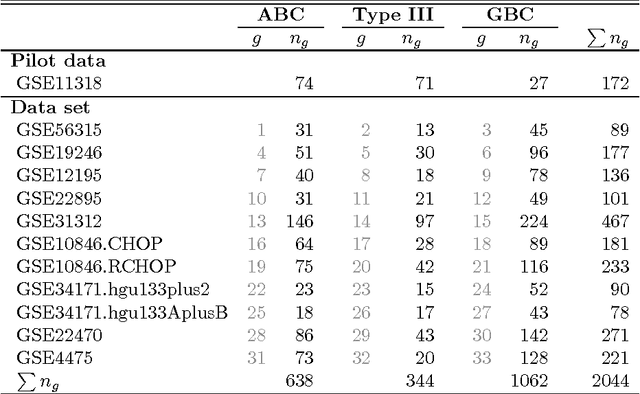
We consider the problem of jointly estimating multiple precision matrices from (aggregated) high-dimensional data consisting of distinct classes. An $\ell_2$-penalized maximum-likelihood approach is employed. The suggested approach is flexible and generic, incorporating several other $\ell_2$-penalized estimators as special cases. In addition, the approach allows for the specification of target matrices through which prior knowledge may be incorporated and which can stabilize the estimation procedure in high-dimensional settings. The result is a targeted fused ridge estimator that is of use when the precision matrices of the constituent classes are believed to chiefly share the same structure while potentially differing in a number of locations of interest. It has many applications in (multi)factorial study designs. We focus on the graphical interpretation of precision matrices with the proposed estimator then serving as a basis for integrative or meta-analytic Gaussian graphical modeling. Situations are considered in which the classes are defined by data sets and/or (subtypes of) diseases. The performance of the proposed estimator in the graphical modeling setting is assessed through extensive simulation experiments. Its practical usability is illustrated by the differential network modeling of 11 large-scale diffuse large B-cell lymphoma gene expression data sets. The estimator and its related procedures are incorporated into the R-package rags2ridges.